Abstract
PGD2 is a key mediator of allergic inflammatory diseases that is mainly synthesized by mast cells, which constitutively express high levels of the terminal enzyme involved in PGD2 synthesis, the hematopoietic PGD synthase (H-PGDS). In this study, we investigated whether eosinophils are also able to synthesize, and therefore, supply biologically active PGD2. PGD2 synthesis was evaluated within human blood eosinophils, in vitro differentiated mouse eosinophils, and eosinophils infiltrating inflammatory site of mouse allergic reaction. Biological function of eosinophil-derived PGD2 was studied by employing inhibitors of synthesis and activity. Constitutive expression of H-PGDS was found within nonstimulated human circulating eosinophils. Acute stimulation of human eosinophils with A23187 (0.1–5 μM) evoked PGD2 synthesis, which was located at the nuclear envelope and was inhibited by pretreatment with HQL-79 (10 μM), a specific H-PGDS inhibitor. Prestimulation of human eosinophils with arachidonic acid (10 μM) or human eotaxin (6 nM) also enhanced HQL-79–sensitive PGD2 synthesis, which, by acting on membrane-expressed specific receptors (D prostanoid receptors 1 and 2), displayed an autocrine/paracrine ability to trigger leukotriene C4 synthesis and lipid body biogenesis, hallmark events of eosinophil activation. In vitro differentiated mouse eosinophils also synthesized paracrine/autocrine active PGD2 in response to arachidonic acid stimulation. In vivo, at late time point of the allergic reaction, infiltrating eosinophils found at the inflammatory site appeared as an auxiliary PGD2-synthesizing cell population. Our findings reveal that eosinophils are indeed able to synthesize and secrete PGD2, hence representing during allergic inflammation an extra cell source of PGD2, which functions as an autocrine signal for eosinophil activation.
Mast cells and eosinophils, two of the principal effector cell types activated at the sites of allergic inflammation, are major participants in the pathogenesis of asthma and other forms of allergic disorders (1, 2). Mast cells and eosinophils have the potential to generate and release diverse lipid mediators that are critical to the development and perpetuation of allergic inflammation, including PGs and leukotrienes derived from the oxidative metabolism of arachidonic acid (AA). Both mast cells and eosinophils express the sole leukotriene (LT)C4-synthesizing enzyme, named LTC4 synthase, and are major cell sources of cysteinyl LTs (cysLTs). LTC4 and its extracellular derivatives, LTD4 and LTE4, have many well-recognized functions as mediators of the allergic response, causing bronchoconstriction, mucous hypersecretion, increased microvascular permeability, bronchial hyperresponsiveness, and eosinophil infiltration and activation (3, 4). Different from LTC4-synthesizing capabilities, specific prostanoids produced by mast cells and eosinophils appear to differ according to their differential expression of prostanoid-synthesizing terminal isoenzymes (5, 6). Concerning prostanoids with allergy-relevant functions, mast cells are considered the predominant cellular domain of hematopoietic PGD2 synthase (H-PGDS) among resident and recruited cells in allergic inflammatory tissues (7); PGD2 is also the major prostanoid produced by mast cells (8). Indeed, PGD2 and its metabolites have been proposed to be selective markers for mast cell activation in vivo (9–11). But recently, the dogma that mast cells are the single PGD2 source in allergic inflammatory conditions has been challenged. New findings unveiled that although PGD2 synthesis seems to be primarily controlled by allergy-relevant cells, it is not restricted to mast cells. Among these additional PGD2-synthesizing cellular sources are the following: 1) both direct and indirect endothelium-mediated generation of PGD2 (12, 13); 2) Th2 lymphocyte capability of synthesizing small, yet significant, amounts of PGD2 (14); 3) skin dendritic cells as supplier of PGD2 with roles in skin inflammation (15); 4) host defense-related PGD2 synthesis by activated macrophages (16, 17); and 5) basophil-driven PGD2 release (18). What about eosinophils? Among prostanoids, it is well accepted that eosinophils are producers of the putative mediators, thromboxane A2 and PGE2, rather than mediators of allergic reactions. It is presumed that the lack of PGD2-synthesizing capability by eosinophils relies on anecdotal evidence of no H-PGDS expression within eosinophils. Nevertheless, whereas some indications of PGD2-synthesizing activity may exist (19–21), definitive demonstration that eosinophils can generate PGD2 is still lacking.
Understanding the mechanisms governing PGD2 synthesis, including the identification of specific PGD2-producing cells, is important, as PGD2 has emerged as a key mediator of the pathogenesis of allergic diseases. PGD2 recruits and activates eosinophils, as well as basophils and Th2 lymphocytes (22–24). In vivo, PGD2 administration in human volunteers or animals imitates a variety of allergic features (25, 26). PGD2 effects are mediated by the activation of the two known PGD2 receptors, namely D prostanoid receptor (DP)1 and DP2 (also known as chemoattractant receptor-homologous molecule expressed on Th2 cells) (22, 27–29). Eosinophils coexpress both the classic DP1 receptors coupled to adenylyl cyclase, as well as pertussis toxin-sensitive DP2 (23). It has been shown that PGD2 ability to activate eosinophils may be determined by a balance between these DP1- versus DP2-driven opposing downstream signaling pathways (e.g., PGD2-induced eosinophil chemotaxis) (23, 30, 31), but alternatively may well be dependent on an initially unexpected DP1/DP2 cooperative effect (e.g., PGD2-elicited enhanced LTC4 synthesis by eosinophils) (32). The appeal of PGD2 as a therapeutic target in allergic diseases, such as asthma, can be promptly attested by the rapid development of selective pharmacological tools to examine the proallergic contributions of these two receptors. Of note, because a variety of prostanoid molecules, including PGD2 metabolites, PGF2, and 11-dehydro-thromboxane B2, are capable of activating DP2 (22, 33–38), one can hypothesize physiopathological outcomes of activation of PGD2 receptors even in the absence of PGD2 production. However, the concentrations of PGD2 are indeed elevated in a variety of chronic allergic tissues, including in the nasal mucosa of allergic rhinitis (39), the airways of asthmatics (40, 41), and the skin of patients with atopic dermatitis (42). Although in these conditions PGD2 synthesis is portrayed as a predominantly mast cell-derived product (41), little is known about the alternative and complementary cell sources of PGD2.
Our study reports that, upon proper stimulation, both human and mouse eosinophils can produce significant amounts of biologically relevant PGD2. PGD2 intracellular synthesis within eosinophils was catalyzed by eosinophil-expressed H-PGDS and led to PGD2 receptor-mediated paracrine/autocrine functions, contributing to eosinophil activation.
Materials and Methods
Animals
Swiss and BALB/c mice of 16–20 g from both sexes were used. The animals were obtained from the Oswaldo Cruz Foundation breeding unit (Rio de Janeiro, Brazil). The protocols were approved by the Oswaldo Cruz Foundation Animal Welfare Committee.
Allergic pleurisy in sensitized mice
As previously described (43), mice were sensitized with a s.c. injection (0.2 ml) of OVA (50 μg) and Al(OH)3 (5 mg) in a 0.9% NaCl solution (saline) at days 1 and 7. Allergic challenge was performed at day 14 by means of an intrapleural injection of OVA (12 μg/cavity; 0.1 ml). Control animals received vehicle (saline; 0.1 ml). The mice were euthanized by CO2 inhalation 48 h after challenge. The pleural cavities were rinsed with 500 μl Ca2+/Mg2+-free HBSS (pH 7.4; HBSS−/−).
Pleural eosinophil counts
Total leukocyte counts were performed using a Neubauer chamber under an optical microscope, after dilution with Turk fluid (2% acetic acid). Differential counts of mononuclear cells, neutrophils, and eosinophils were performed under an oil immersion objective using cytopins (Cytospin 3; Shandon, Pittsburgh, PA) stained by the May-Grunwald-Giemsa method. Counts are reported as eosinophils per cavity.
Isolation of human blood eosinophils
Peripheral blood was obtained with informed consent from normal donors. Briefly, following dextran sedimentation and Ficoll gradient steps, eosinophils were isolated from contaminating neutrophils by negative immunomagnetic selection using the EasySep system (StemCell Technologies), which includes Abs against human CD2, CD3, CD14, CD16, CD19, CD20, CD36, CD56, CD123, and glycophorin A coupled to magnetic particles (cell purity ∼99%; cell viability ∼95%) (32). The protocol was approved by ethical review boards of both the Federal University of Rio de Janeiro and the Oswaldo Cruz Foundation (Rio de Janeiro, Brazil).
In vitro stimulation of human eosinophils
Purified human eosinophils at 2 × 106 cells/ml in Ca2+/Mg2+ HBSS (HBSS+/+; pH 7.4) were incubated with A23187 (0.1–5 μM; Sigma-Aldrich) for 15 min in a water bath (37°C). Alternatively, eosinophils were stimulated with AA (10 μM; Cayman Chemicals), human recombinant eotaxin (also known as CCL11 or eotaxin-1; 6 nM; R&D Systems), macrophage migration inhibitory factor (MIF; 50 ng/ml; R&D Systems), platelet-activating factor (PAF; 1 μM), or PGD2 (25 nM) in HBSS−/− for 1 h. To enable detection of released PGD2 or LTC4 by enzyme immunoassay (EIA), AA- or eotaxin-stimulated eosinophils were also challenged with 0.1 μM A23187 (Sigma-Aldrich) for an additional 15 min in HBSS+/+. Each condition was repeated at least three times with eosinophils purified from different donors.
In vitro eosinophil differentiation from mouse bone marrow cells
With slight modifications, eosinophils were differentiated in vitro from mouse bone marrow cells, as previously described (44). Briefly, bone marrow cells were collected from femurs and tibiae of wild-type BALB/c mice with RPMI 1640 (Sigma-Aldrich) containing 20% FBS. After RBC lysis, cells were cultured at 106 cells/ml in RPMI 1640 containing 20% FBS (VitroCell), 100 IU/ml penicillin, 10 μg/ml streptomycin, 2 mM glutamine (Sigma-Aldrich), 1 mM sodium pyruvate (Sigma-Aldrich), 100 ng/ml stem cell factor (PeproTech), and 100 ng/ml FLT3 ligand (PeproTech) from days 0 to 4. On day 4, the stem cell factor and FLT3 ligand were replaced with IL-5 (10 ng/ml; Peprotech). On day 14, eosinophils were enumerated (purity ≥75%), resuspended in HBSS−/− (2 × 106 cells/ml), and stimulated with AA (10 μM; Cayman Chemicals), murine recombinant eotaxin (6 nM), PAF (1 μM), or PGD2 (25 nM).
Treatments
For in vitro studies, human or mouse eosinophils in HBSS−/− were pretreated for 30 min with the H-PGDS inhibitor HQL-79 (10 μM), DP1 receptor antagonist BWA868c (200 nM), or chemoattractant receptor-homologous molecule expressed on Th2 cells receptor antagonists Bay-u3405 (200 nM) and Cay 10471 (200 nM), or concomitantly with Abs against PGD2 (10 μl; all from Cayman Chemicals) or its isotype control. Notably, pretreatments did not modify the eosinophil basal lipid body content, nor did they affect eosinophil viability (∼90%) (data not shown). For in vivo assays using the pleurisy model, animals were pretreated with i.p. injections of HQL-79 (1 mg/Kg) 30 min before allergic challenge.
Stock solutions of stimuli and inhibitors were prepared in HBSS−/− containing 0.1% endotoxin-free OVA, aliquoted, and stored at −20°C. Specifically, concentration of HQL-79 stock solution was 5 mM in DMSO (DMSO final concentration during cell incubations was 0.02% and had no effect on eosinophils). A23187, BWA868c Bay-u3405, and Cay 10471 were also diluted in DMSO, whereas AA, PAF, and PGD2 were diluted in ethanol. The final vehicle concentration was <0.01% and had no effect on eosinophils.
H-PGDS immunolocalization
Human eosinophils (2 × 105 cells) were cytocentrifuged (500 rpm, 5 min) onto glass slides, and fixed in 2% paraformaldehyde for 10 min. After washing (3 × 10 min) with 0.05% saponin (Sigma-Aldrich) containing 1% BSA (Sigma-Aldrich) in HBSS−/−, the slides were incubated for 30 min with rabbit polyclonal antiserum anti–H-PGDS (Cayman Chemicals) or with normal rabbit serum, washed with saponin/BSA, incubated for 30 min with Alexa Fluor 488-labeled goat anti-rabbit IgG Ab (Molecular Probes), washed with saponin, washed with HBSS−/−, and then an aqueous medium containing DAPI (Vector Laboratories). The images were obtained using an Olympus BX51 fluorescence microscope equipped with a Plan Apo original magnification ×100 1.4 Ph3 objective and an Olympus 72 digital camera (Olympus Optical) in conjunction with CellF Imaging Software for Life Science Microscopy (Olympus Life Science Europe GMBH). The images were edited using Adobe Photoshop 5.5 software (Adobe Systems).
Quantification of eicosanoids
PGD2, PGE2, or cysLTs found in cell-free pleural fluid and eosinophil supernatants were measured by commercial EIA kits, according to the manufacturer´s instructions (Cayman Chemicals).
EicosaCell for intracellular PGD2 immunodetection
By using EicosaCell (45) to detect intracellular PGD2 at its sites of synthesis, in vitro stimulated eosinophils (in HBSS+/+) or pleural cells were mixed with an equal volume of 1-ethyl-3-(3-dimethylamino-propyl) carbodiimide (EDAC) solution (0.2% in HBSS−/− containing 1% BSA). After a 15-min incubation at 37°C with EDAC, eosinophils were washed with HBSS−/−, cytospun onto glass slides, incubated with HBSS−/− with 1% BSA for 30 min, and then incubated with rabbit anti-PGD2 Abs (Assay Designs) overnight. Notably, PGD2 immunodetection was also achieved when another anti-PGD2 Ab (Cayman Chemicals) was employed (data not shown). The anti-adipose differentiation-related protein (ADRP) Ab was also added overnight to distinguish cytoplasmic lipid bodies within eosinophils. The cells were washed with HBSS−/− containing 1% BSA (3× 10 min) and then incubated with DyLight488 anti-mouse IgG and DyLight549 anti-guinea pig secondary Abs (Jackson ImmunoResearch Laboratories) for 1 h. EicosaCell images were obtained using a LEICA TCS–SP5 confocal fluorescence microscope, equipped with an original magnification ×63 objective in conjunction with LAS-AF 2.2.0 Software.
Alternatively, to detect PGD2-synthesizing eosinophils by flow cytometry, after incubation with EDAC, pleural fluid cells were washed with HBSS−/−, incubated overnight with rabbit anti-PGD2 Abs, washed with HBSS−/−, and then incubated with DyLight488 anti-mouse IgG and Ab anti–SiglecF-PE (or isotype-PE) (from eBioscience) for 30 min. After washings, cells were analyzed by flow cytometry in a FACSCalibur (BD Biosciences) flow cytometer.
RT-PCR
mRNA was extracted from 106 nonstimulated or AA-stimulated human eosinophils, according to the manufacturer's protocol (RNeasy kit; Qiagen, Germantown, MD). cDNA synthesis and RT-PCR conditions followed standard protocols. Primer sequences for human H-PGDS were the same as previously published (46) and for human β-actin were 5′-GACAGGATGCAGAAGGAGAT-3′ and 5′-TGTGTGGACTTGGGAGAGGACT-3′ (based on GenBank sequence X00351).
Lipid body staining and enumeration
Cytospun cells, although still moist, were fixed in 3.7% formaldehyde (diluted in HBSS−/−), rinsed in 0.1 M cacodylate buffer (pH 7.4), stained with 1.5% OsO4 for 30 min, rinsed in dH2O, immersed in 1.0% thiocarbohydrazide for 5 min, rinsed in 0.1 M cacodylate buffer, restained with 1.5% OsO4 for 3 min, rinsed in distilled water, and then dried and mounted. The cell morphology was observed, and the lipid bodies were enumerated by light microscopy. Fifty consecutively scanned eosinophils were evaluated in a blinded fashion by more than one individual, and the results were expressed as the number of lipid bodies per eosinophil.
Statistical analysis
The results were expressed as mean ± SEM and were analyzed statistically by means of ANOVA, followed by Student–Newman–Keuls test, with the level of significance set at p < 0.05.
Results
Human circulating eosinophils are enzymatically competent cells for prompt synthesis of PGD2
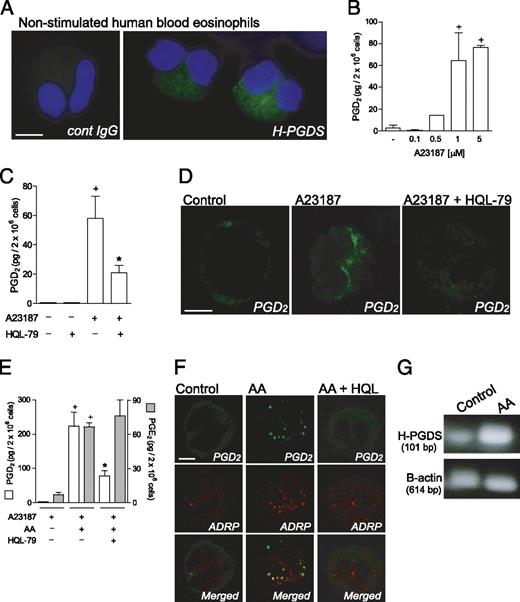
Analyses by fluorescence microscopy demonstrated that H-PGDS is constitutively expressed by human circulating eosinophils freshly isolated from healthy donors. As shown in Fig. 1A, H-PGDS labeling within nonstimulated human blood eosinophils displayed a cytoplasmic staining pattern. No immunoreactivity was detected within eosinophils incubated with control normal rabbit serum (Fig. 1A). Thus, in addition to H-PGDS expression within activated human eosinophils, such as those recruited to nasal mucosa of patients with allergic rhinitis (21) or polyps of chronic rhinosinusitis patients (20), resting eosinophils also contained detectable amounts of cytoplasmic H-PGDS.
It is now well recognized that merely the expression of eicosanoid-forming enzymes does not determine successful eicosanoid synthesis (for a review, see Ref. 47). To investigate whether eosinophil H-PGDS could couple to upstream prostanoid-synthesizing enzymes and mount an active PGD2-synthesizing machinery, human blood eosinophils were stimulated with calcium ionophore A23187. As shown in Fig. 1B, A23187 dose dependently (0.1–5 μM) elicited acute (within 15 min) secretion of PGD2 from eosinophils. The pretreatment with a selective inhibitor of H-PGDS, HQL-79 (10 μM), inhibited PGD2 synthesis/release induced by A23187 (5 μM; Fig. 1C), but failed to attenuate concomitant LTC4 secretion (data not shown), therefore validating both PGD2 detection by EIA and the specificity of HQL-79 treatment, although evidencing that eosinophils can rapidly organize an effective H-PGDS–dependent PGD2 synthesis.
EicosaCell, an immunoassay that immobilizes the newly formed eicosanoid at its intracellular synthesizing compartments (45), confirmed eosinophil’s ability to synthesize PGD2. As illustrated in Fig. 1D, nonstimulated eosinophils exhibited no immunofluorescent staining for PGD2. In contrast, virtually all eosinophils (>90%) activated for 15 min with 0.1 μM A23187 yielded intense and localized immunofluorescent staining for PGD2 with a perinuclear localization, a well-established eicosanoid-forming site (Fig. 1D). The specificity of EicosaCell staining for PGD2 was ascertained, because there was no immunostaining when an isotype control Ab was used (data not shown), and mainly because HQL-79, which blocks H-PGDS activity, completely abolished PGD2 staining in A23187-stimulated eosinophils (Fig. 1D). Although A23187-driven activation easily elicits the enzymatic pathways for eicosanoid production, it is supraphysiologic and may not depict physiopathologic mechanisms of PGD2 synthesis. Therefore, the unsaturated fatty acid AA, which functions as both substrate and physiologic stimulus of eicosanoid synthesis (48, 49), was employed.
As illustrated in Fig. 1E, AA (10 μM) very effectively primed eosinophils for increased PGD2 release in response to a submaximal, 0.1 μM concentration of A23187 (Fig. 1E). AA-stimulated eosinophils released ∼200 fold as much PGD2 as eosinophils challenged with 0.1 μM A23187 alone. Moreover, HQL-79 inhibited AA-primed PGD2 production by eosinophils, while failing to affect concurrent synthesis of PGE2 (Fig. 1E). This inhibition confirmed the specificity of HQL-79 and reinforced the role of H-PGDS in AA-driven PGD2 synthesis by eosinophils. Moreover, it is not surprising that whereas the amounts of secreted PGD2 are not that different from those picograms of PGE2 found in the supernatant of AA-stimulated A23187-challenged eosinophils (Fig. 1E), under the same conditions eosinophils synthesize larger amounts (nanograms) of LTC4.
Although minute quantities of PGD2 produced by eosinophils stimulated only with AA were not sufficient to be detectable in supernatants by EIA (data not shown), it was intracellularly detected by EicosaCell. As shown in Fig. 1F, AA (10 μM) was able to trigger within human eosinophils PGD2 synthesis, which was immunodetected within 30 min of stimulation. A detailed analysis revealed that part of newly synthesized PGD2 (green labeling) was in a punctate cytoplasmic pattern proximate to, but separate from, the nucleus and fully consistent in size and form with eosinophil lipid bodies. The specific compartmentalization of newly formed PGD2 within eosinophil lipid bodies was ascertained by the colocalization with ADRP, a protein marker of lipid bodies (Fig. 1F, red labeling). Virtually no PGD2 immunolabeling was observed within nonstimulated eosinophils (Fig. 1F), whereas ∼91% of eosinophils stimulated with AA exhibited lipid body-localized staining for immunoreactive PGD2, which was fully inhibitable by HQL-79 (Fig. 1F). Collectively, these data evidence the role of H-PGDS in AA-induced PGD2 synthesis by eosinophils and ascertain the specificity of the PGD2 immunolabeling. Therefore, newly formed lipid bodies of AA-stimulated eosinophils are inducible and enzymatically skilled organelles for effective PGD2 synthesis. Concurrently, RT-PCR analysis showed that nonstimulated human circulating eosinophils express H-PGDS mRNA, which may represent a potential target for an AA-driven priming effect on PGD2 production by eosinophils because, in parallel, AA stimulation increased levels of H-PGDS message (Fig. 1G).
Eotaxin elicits the synthesis of biologically active PGD2
Eotaxin, a key mediator in the development of allergic eosinophilia that is known by its potent eosinophilotactic activity, has recently emerged as a potent mediator of eosinophil activation, with the particular ability to enhance LTC4 synthesis by eosinophils (50). In this study, we showed that, aside from activating 5-lipoxygenase pathways, eotaxin is also able to control cyclooxygenase-driven prostanoid synthesis within eosinophils. As shown in Fig. 2A, the prestimulation with eotaxin (6 nM) effectively primed eosinophils for HQL-79–sensitive enhanced PGD2 release in response to suboptimal concentration of A23187. Eotaxin-prestimulated eosinophils released ∼13-fold as much PGD2 as did eosinophils challenged with A23187 alone (0.1 μM). Although eosinophils stimulated with eotaxin alone released levels of PGD2 that are not detectable by EIA (data not shown), our findings demonstrate eotaxin ability to initiate H-PGDS–driven PGD2-synthesizing machinery within eosinophils. To definitively establish eotaxin ability to trigger H-PGDS–driven PGD2-synthesizing machinery within eosinophils, more sensitive EicosaCell assay was employed with eotaxin stimulation of either human or mouse eosinophils. As shown in Fig. 2D and 2F, eotaxin triggered HQL-sensitive lipid body-compartmentalized PGD2 synthesis within human and mouse eosinophils. In human cells, immunofluorescence PGD2 was detected as acute as 4 min of stimulation with eotaxin in ∼40% of eosinophils (Fig. 2D), whereas ∼90% of eotaxin-stimulated mouse eosinophils displayed PGD2 immunolabeling (Fig. 2F). Corroborating such cell-primed state of in vitro differentiated mouse eosinophils, eotaxin stimulation per se was able to elicit PGD2 production that was detected even by EIA in cell supernatants (Fig. 2E). It is also noteworthy that any speculation, that other PGD2-synthesizing cells contaminating eosinophil preparations (e.g., basophils) were the actual cell type responsible for PGD2 synthesis, was ruled out by the high percentage of cells in eosinophil preparations synthesizing PGD2. Either in AA-stimulated human eosinophils (Fig. 1E) or eotaxin-stimulated mouse eosinophils (Fig. 2F), by EicosaCell ∼90% of cells were PGD2 immunoreactive.
To characterize whether eotaxin-induced eosinophil-derived PGD2 was a bioactive mediator, by employing HQL-79 as a pharmacological strategy, we evaluated the role of endogenous PGD2 on two parameters of eosinophil functions triggered by eotaxin (6 nM): enhanced LTC4-synthesizing ability and induction of lipid body biogenesis. As shown in Fig. 2B and 2C, eotaxin-induced enhancement of cysLTs production and lipid body biogenesis was blocked by HQL-79. The inhibition of eotaxin-induced eosinophil functions by HQL-79, besides reinforcing that eosinophils do in fact synthesize PGD2, shows that eosinophil-derived PGD2 displays biological activity, which can be very acute (even within 4 min), with allergy-relevant impacts on eosinophil activation.
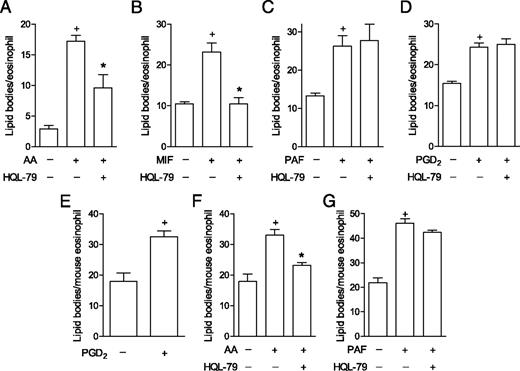
Induction of synthesis of bioactive PGD2 is stimulus specific
As a functional approach to further screen potential physiological stimuli of PGD2 synthesis by eosinophils, lipid body biogenesis, a complex cellular outcome (47) that can be triggered by PGD2 (43), was induced by a variety of stimuli undergoing HQL-79 treatment. As shown in Fig. 3A and 3B, either AA- or MIF-induced biogenesis of cytoplasmic lipid bodies within eosinophils was significantly reduced by HQL-79, indicating that, at least in part, lipid body assembly triggered by these stimuli is dependent on endogenously produced PGD2. Among eosinophil-relevant agonists with a recognized capacity to trigger lipid body biogenesis, PAF and PGD2 itself also trigger the rapid, receptor-mediated assembly of lipid bodies through signaling mechanisms distinct from those of AA and eotaxin (47). In this study, we highlighted such signaling diversity and characterized the eosinophil PGD2-synthesizing ability as a stimulus-specific process, because HQL-79 treatment failed to modify either PAF (Fig. 3C)- or PGD2-elicited lipid body biogenesis (Fig. 3D). Of note, corroborating PAF lack of ability to induce PGD2 synthesis within eosinophils, distinct from eotaxin (Fig. 2D), PAF-stimulated eosinophils fail to mount PGD2-synthesizing machinery (within 4 min or 1 h), because EicosaCell preparations of PAF-stimulated human eosinophils did not display PGD2 immunofluorescence (data not shown).
Mouse eosinophil-derived PGD2 is also an endogenous bioactive molecule
Similar to human eosinophils, mouse eosinophil-derived PGD2 also displayed endogenous stimulatory effects. After establishing that exogenous PGD2 is capable of triggering lipid body biogenesis within mouse eosinophils (Fig. 3E), we demonstrated that endogenous PGD2 released from AA-stimulated (Fig. 3F), but not from PAF-stimulated, mouse eosinophils (Fig. 3G) also participates in subsequent lipid body assembly. Taken together, these findings discard potential unspecific effects of HQL-79 on eosinophils, confirm that eosinophil-derived PGD2 contributes to lipid body assembly in a stimulus-specific fashion, and demonstrate the PGD2-synthesizing properties of mouse eosinophils. Therefore, rather than a species-specific phenomenon, the eosinophil PGD2-synthesizing ability appears to be a broad function displayed by the eosinophil cell type.
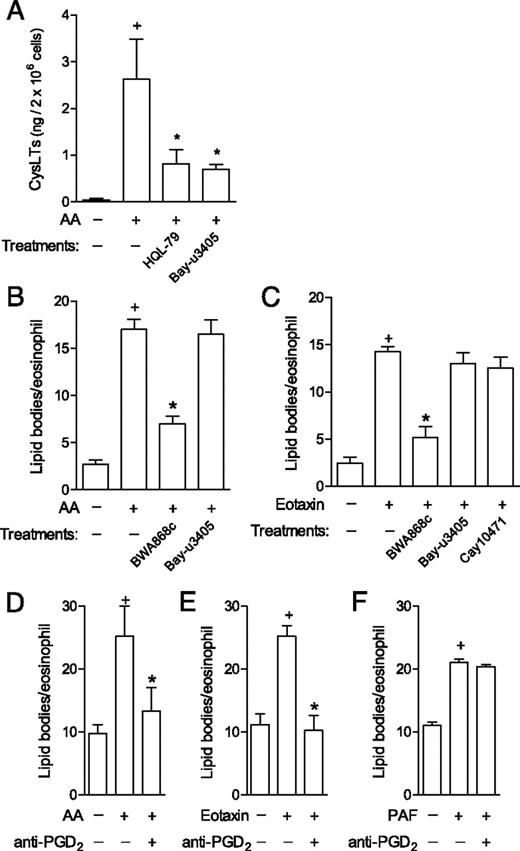
Eosinophil-derived PGD2 triggers eosinophil activation via interaction with specific receptors
We hypothesized that eosinophil-derived PGD2 may regulate AA- and eotaxin-induced eosinophil activation by acting on its specific receptors DP1 and DP2. Pretreatments with HQL-79 or Bay-u3405 (antagonist of the PGD2 receptor DP2) significantly inhibited eosinophil cysLTs production triggered by AA, thus showing that newly synthesized PGD2 functions as an agonist of eosinophil-expressed DP2 receptors under specific stimulation (Fig. 4A). Similarly, lipid body assembly triggered by either AA (Fig. 4B) or eotaxin (Fig. 4C) was significantly reduced when the DP1 receptor was blocked with the selective DP1 antagonist BWA868c, indicating that AA- and eotaxin-stimulated eosinophils release biologically active PGD2, which binds to DP1 receptors expressed on eosinophils to initiate lipid body biogenesis-eliciting signaling. Fig. 4 also shows that pretreatments with antagonists of the PGD2 receptor DP2, Bay-u3405 or Cay10471, as expected, failed to affect AA (Fig. 4B)- or eotaxin-induced lipid body biogenesis (Fig. 4C), confirming that the lipid body biogenic process within eosinophils triggered by exogenous or endogenous PGD2 is controlled selectively by DP1 activation (32).
Endogenous PGD2 displays an autocrine/paracrine, rather than an intracrine, effect on eosinophils
Acting extracellularly, PGD2 has emerged as key paracrine mediator pertinent to asthma and other allergic diseases. However, it is increasingly accepted that eicosanoids, including PGD2, may also display intracrine roles in regulating cell functions. Indeed, signaling evoked by intracellular eicosanoid receptors has been shown in eosinophils and other cells (51–53). To verify whether autocrine/paracrine versus intracrine activity of eosinophil-synthesized PGD2 controls eosinophil activation, intact viable eosinophils were pretreated with anti-PGD2 Abs whose neutralizing activity is excluded from intracellular compartment. Pertinent to PGD2 functions as a paracrine/autocrine mediator of eosinophil activation (Fig. 4), the neutralization of endogenous PGD2 by the anti-PGD2, but not by an isotype control (data not shown), inhibited lipid body biogenesis induced by either AA (Fig. 4D) or eotaxin (Fig. 4E) within human eosinophils. Supporting Ab specificity, anti-PGD2 treatment failed to affect PAF-induced lipid body formation within eosinophils (Fig. 4F). Thus, rather than functioning as an intracrine signal-transducing molecule, eosinophil-derived PGD2 is secreted from eosinophils and then, by acting extracellularly on PGD2 specific receptors, mediates eosinophil activation initiated by AA or eotaxin.
Eosinophils recruited to sites of allergic inflammation function as a late cellular source of PGD2
As activation of infiltrating eosinophils is a critical feature in the pathogenesis of allergic diseases, we hypothesized that activated eosinophils found at sites of allergic inflammation may synthesize and release PGD2. As previously described (43, 54, 55), allergic challenge in actively sensitized mice induces a marked eosinophil recruitment to the pleural cavity in a mouse model of allergic inflammation. Infiltrating eosinophils are not detectable within 1 h, peak at 24 h, are detectable within the pleural space up to at least 96 h after allergic challenge, and are concurrent with a resident population of mast cells and a discrete, but significant accumulation of other mononuclear cells (macrophages and lymphocytes), but no neutrophil is found (54). It is noteworthy that at acute time points (within 1 h) in absence of allergic eosinophilia, pleural inflammatory fluid presents a significant increase in PGD2 amounts (from 13 ± 10 to 61 ± 12 ng/cavity of PGD2 in saline- versus OVA-challenged mice, respectively; p ≤ 0.05; n = 3); such synthesis can be attributed to resident mast cells. Notably, 48-h–related pleural eosinophilia (Fig. 5A), which is insensitive to HQL-79, parallels an increased pleural level of PGD2 (Fig. 5B). Such delayed allergen-elicited PGD2 production appears to depend on H-PGDS activity, because HQL-79 impairs it (Fig. 5B). To identify the cell source of delayed pleural PGD2, EicosaCell preparations of pleural leukocytes recovered from allergic inflammatory sites were immunolabeled with anti-PGD2. As shown in Fig. 5C (left panel), virtually all eosinophils infiltrating the pleural space yielded focal immunofluorescent staining for PGD2. Of note, in EicosaCell preparations, the eosinophil population was readily identified by visual inspection of nucleus morphology, because as defined by direct counting of eosin-stained cells, the eosinophils (∼35% of total pleural cells) were the single polymorphonuclear cell type found infiltrating allergic site of inflammation. The specificity of PGD2 immunostaining within recruited eosinophils was validated because of the following: 1) leukocytes from sensitized, nonchallenged mice exhibited no PGD2 staining; 2) pretreatment with HQL-79 completely abolished PGD2 staining within infiltrating eosinophils (Fig. 5C, right panel); and 3) mononuclear cells (e.g., macrophages), also found in the pleural space after an allergic challenge in sensitized mice, did not show any staining for intracellular PGD2 (Fig. 5C, left panel) at this time point. Even though mast cells are the professional PGD2-synthesizing cells, at late time points mast cell contribution to PGD2 generation appears to be very limited (Fig. 5C, left panel, arrows). The ability of eosinophils recruited to site allergic inflammation to produce PGD2 was further confirmed by analyzing EicosaCell preparations by flow cytometry (Fig. 5D). To study eosinophils, we employed double immunolabeling by anti-SiglecF, an effective approach at detecting eosinophils in mixed cell populations (56). Indeed, as expected, the SiglecF staining yielded results that matched visual inspection of eosin-stained cytospin preparations by specifically labeling eosinophilic polymorphonuclear cells (Fig. 5D, left panel), immunodetecting 32.1 ± 6.2% of cells infiltrating allergic inflammatory space as SiglecF+ eosinophils (n = 5). Specifically analyzing these SiglecF+ cells in Eicosacell preparations of allergic reaction by flow cytometry (Fig. 5D, right panel), we extended our microscopic findings by demonstrating that all recruited SiglecF+ eosinophils synthesized PGD2, as evidenced by an HQL-79–sensitive uniformly positive, unimodal pattern of intracellular PGD2 immunoreactivity (Fig. 5D). Collectively, our data implicate infiltrating eosinophils as an additional cell population responsible for maintenance of PGD2 production during allergic inflammatory reactions.
Discussion
PGD2 is a key lipid mediator of allergic airway inflammation that is released following allergen exposure in patients with asthma (40, 41). Because PGD2 modulates key aspects of this prevalent pathology, PGD2 has emerged as a major mediator of allergic inflammatory disorders, and, therefore, is an interesting target for anti-allergic treatments. Among PGD2-driven asthma-relevant actions are the synthesis of cysLTs, as well as both recruitment and subsequent activation of eosinophils, which is one of the principal cell types recruited to and activated at sites of allergic inflammation.
PGD2 is a major cyclooxygenase pathway product of mast cells, which are acknowledged as a key cell population providing PGD2 within inflammatory sites of allergic reactions. This view is mostly based on a rather restricted expression of the limiting PGD2-forming enzyme H-PGDS within mast cells. Extending the findings of an earlier study that evaluated human tissue eosinophils found infiltrating mucosa of patients with allergic rhinitis (21) or polyps of chronic rhinosinusitis patients (20), H-PGDS expression (both at mRNA and protein levels) was detected within human circulating eosinophils freshly isolated from healthy donors, thus indicating that, in addition to mast cells, eosinophils could also contribute to PGD2 synthesis in allergic inflammatory sites. However, it is now well established that the successful production of PGD2, or any other eicosanoid, is not merely determined by the proper expression of restrictive enzymes. In addition, it requires AA availability, the presence of all other relevant protein/enzymes, coordinated phosphorylation of some enzymes, correct spatial assembly of enzymatic complexes, and regulated intracellular compartmentalization of these complexes (for a review, see Ref. 47). By simply finding H-PGDS within eosinophils, it cannot be ascertained whether these cells are capable of mounting a successful PGD2 production and, consequently, contribute to allergy-related elevated PGD2 generation. Therefore, our main aim in this study was to prove that, as in mast cells, eosinophil H-PGDS could couple to an active prostanoid-synthesizing machinery at specific, intracellular compartments within eosinophils, which would generate and release bioactive PGD2. By employing several experimental techniques, we attest that both human and mouse eosinophils are indeed able to synthesize PGD2 because of the following: 1) PGD2 was detected within supernatants of purified human eosinophils stimulated in vitro with A23187; 2) AA and eotaxin were able to upregulate PGD2 production/release by eosinophils; 3) AA- and eotaxin-primed, as well as A23187-induced, PGD2 production/release were inhibited by HQL-79; 4) A23187- and AA-induced, HQL-79–sensitive PGD2 synthesis was differentially compartmentalized within the perinuclear membrane and lipid bodies, respectively; and 5) functional assays show that AA- and eotaxin-induced eosinophil activation were inhibited by pretreatments with HQL-79, Ab anti-PGD2, or receptor antagonists of DP1 and DP2. More specifically, these data show that such PGD2 synthesis by eosinophils is a H-PGDS–dependent event that culminates in an extracellular release of a biologically active PGD2, which displays autocrine/paracrine activities on eosinophils via the activation of its specific receptors, DP1 and DP2.
What about eosinophil contribution to allergic airway inflammation as a PGD2 source following allergen exposure? To definitively demonstrate that eosinophils also play such an additional role in allergic inflammatory reactions by providing PGD2, we employed a direct strategy for the in situ immunolocalization of intracellular PGD2 to identify the cell population responsible for PGD2 production in a mouse model of allergic inflammation that displays increased levels of PGD2 at later time points. Eosinophils recruited to the inflammatory site, which also had concurrent macrophage and mast cell populations, were the predominant cell type generating PGD2, thus challenging the prevailing notion of mast cells as the single PGD2 cell source during allergic reactions and placing eosinophils as responsible for continued production of PGD2.
Eosinophil-derived, biologically active PGD2 may modulate eosinophil activation in sites of allergic inflammation. PGD2-driven eosinophil activation during allergic airway inflammation is known to elicit LTC4 production by eosinophil themselves (43). Therefore, even at minute amounts and in part due to its autocrine feature, such eosinophil-derived PGD2 activity emerges as a key upregulatory mechanism for the local generation of proallergic mediators. Considering both our findings and the disappointing clinical trial results of DP1 antagonist laropiprant in asthmatics and allergic rhinitis patients (57), it appears that therapies targeting PGD2 synthesis, rather than receptor antagonism, may display superior beneficial outcomes. Therefore, our data, in addition to reinforcing the notion of eosinophils as major effector cells of allergic disorders, identify the PGD2-synthesizing property of eosinophils as a novel alternative target for anti-allergic therapies.
Footnotes
This work was supported by Conselho Nacional de Pesquisas and Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro from Brazil and National Institutes of Health Grants AI020241, AI051645, and AI022571 (to P.F.W.).
Abbreviations used in this article:
References
Disclosures
The authors have no financial conflicts of interest.