

Abstract
Chlamydia pneumoniae is an obligate intracellular human pathogen causing diseases such as pneumonia, bronchitis, and pharyngitis. Because of its intracellular replication, cell-mediated immune responses are needed to mediate successful defenses of the host. Because dendritic cells play a central role in linking innate immunity and Ag-specific cell-mediated immune responses we asked whether dendritic cells are activated upon contact with C. pneumoniae and whether known Toll like receptors (TLR) are involved in this process. Here we show that C. pneumoniae was taken up by bone marrow-derived murine dendritic cells. Ingested C. pneumoniae appeared to be unable to develop mature inclusion inside dendritic cells. Furthermore, upon contact with C. pneumoniae dendritic cells were potently stimulated because NF-κB was activated and translocated to the nucleus, cytokines like IL-12p40 and TNF-α were secreted, and expression of MHC class II molecules, CD40, CD80, and CD86 was up-regulated. Importantly, secretion of cytokines as well as translocation of NF-κB were dependent on the presence of TLR2 and independent from TLR4 with the exception of IL-12p40 secretion, which was attenuated in the absence of either a functional TLR2 or 4. In conclusion, we show here that recognition of the Gram-negative bacterium C. pneumoniae depends largely on TLR2 and only to a minor extent on TLR4.
The bacterium Chlamydia pneumoniae is an obligate intracellular organism able to infect several cell types like epi- and endothelial cells, smooth muscle cells, and macrophages (1, 2, 3, 4). Target cells are infected by metabolically inactive elementary bodies that develop subsequently into reticular bodies in vacuoles of permissive cells. Abs specific for C. pneumoniae can be detected in the serum of up to 70% of healthy human beings implying that most individuals had contact with these organisms (5). Human diseases attributed to C. pneumoniae include atypical pneumoniae, bronchitis, otitis media, and perhaps arteriosclerosis (5). Structurally, C. pneumoniae displays a cell wall similar to that of Gram-negative bacteria (6). However, two important differences exist. First, despite the presence of all necessary genes for the synthesis of peptidoglycan, this structure could hardly be detected in the chlamydial cell wall by biochemical methods (7, 8, 9). Second, although the outer membrane of Chlamydia contains endotoxin, its structure differs from that of classical endotoxins. Data obtained from structural analysis of C. trachomatis L2 endotoxin show that chlamydial endotoxin contains only five instead of six fatty acid chains, and these chains consist of 14–20 instead of 12–14 C atoms in comparison to highly active endotoxin (10, 11). These structural differences appear to contribute to the weak ability of chlamydial endotoxin to stimulate innate immune cells shown to be ∼100 times weaker compared with endotoxin from Salmonella minnesota (12). Perhaps, Chlamydia minimize the induction of innate immune responses to establish successfully the intracellular replication cycle.
In contrast C. pneumoniae is able to trigger innate as well as adaptive immune responses. For example, macrophages are triggered to release a number of proinflammatory cytokines including TNF-α, IL-1, IL-6, and IL-8 (3, 13). Macrophages also restrict the intracellular replication cycle of C. pneumoniae in that the formation of fully developed reticular bodies is blocked and only smaller inclusions prevail, probably because the fusion of chlamydial vacuoles with lysosomes is not inhibited in contrast to permissive HeLa cells (14, 15). Studies in CD4- or CD8-deficient as well as SCID mice revealed that CD4+ and CD8+ T cells are important for the clearance of C. pneumoniae (16). Clearance seems to be dependent on IFN-γ and NO, because IFN-γ receptor as well as inducible NO synthase (iNOS)2-deficient mice are very susceptible to infection with C. pneumoniae (16). These findings also imply that dendritic cells must play an important role for the clearance of C. pneumoniae from the host.
Mature dendritic cells, also termed professional APCs, are able to present minute amounts of Ag to responsive T cells because they express MHC molecules as well as costimulatory molecules like CD40, CD80, and CD86 with high density (17, 18, 19). In their immature stage dendritic cells express fewer of these surface molecules but are much more potent in uptake and processing of Ag. Pathogen-derived pattern ligands like endotoxin or CpG DNA are able to activate and mature efficiently immature dendritic cells (20).
Innate immune cells recognize microorganisms through pattern recognition receptors. The receptors involved are surface molecules like CD14, expressed on macrophages, as well as the mannose receptor (21, 22). The family of Toll-like receptors (TLR) with at least nine defined members plays an important role in the recognition process of pathogen-derived pattern ligands (23, 24) and are mammalian homologs of the Drosophila Toll receptor as well as 18-wheeler, which are responsible for the defense against fungi and bacteria, respectively (25, 26). It is still controversial whether TLRs bind directly to bacterial patterns (27). Pathogen-derived pattern structures include endotoxin, lipoteichoic acid, peptidoglycan, cell wall components of mycobacteria like lipoarrabinomannan, zymosan from yeast, as well as bacterial CpG DNA (23, 28, 29, 30, 31). Recognition of endotoxin appears to be mediated predominantly through CD14 and TLR4, whereas TLR2 appears to be involved in the recognition of crude Gram-positive bacterial cell walls, peptidoglycan, lipoarrabinomannan, lipoproteins, and zymosan (23, 28, 29, 30, 32). Recently, TLR9 was shown to play an important role in the activation of innate immune cells by bacterial CpG DNA (24). As expected from the results described above, the recognition of complete Gram-positive or Gram-negative bacteria is dependent on TLR2 or TLR4, respectively (33, 34). In vivo TLR4-mutant or TLR2-deficient mice display a higher susceptibility to Salmonella typhimurium or Staphylococcus aureus, respectively (35, 36). Main intracellular signal pathways of both TLR2 and TLR4 seem to merge at the level of the adaptor molecule MyD88 because MyD88-deficient mice are unable to recognize the components described above (37, 38, 39, 40).
This study was performed to analyze the response of murine dendritic cells derived from bone marrow (BMDDC) upon contact with C. pneumoniae. We explored whether BMDDC restrict chlamydial development and change the expression profile of important costimulatory molecules, and we analyzed whether proinflammatory cytokines are secreted. Using TLR4-mutant and TLR2-deficient mice, we also investigated whether the responses of BMDDC toward C. pneumoniae were dependent on these molecules known to be involved in host recognition of microbial patterns.
Materials and Methods
Strains of mice
BALB/c and C57BL/6 mice were purchased from Harlan Winkelmann (Borchen, Germany), C3H/HeJ and C3H/HeN mice were obtained from Charles River (Sulzfeld, Germany). TLR2-deficient mice were donated by Tularik (South San Francisco, CA) and bred in our own animal facility. All mice were kept under specific pathogen-free conditions.
The generation and characterization of TLR2-deficient mice is described by Kirschning et al. (C. Kirschning, G. Meng, R. Schwandner, R. Dziarski, H. Wagner, and K. Pfeffer, manuscript in preparation). Briefly, the 3′ end of the extracellular domain as well as the major part of the transmembrane region of TLR2 were deleted and replaced by gene targeting with a neomycin resistance cassette oriented in reverse direction. Homologous recombination was verified by Northern blot and genomic PCR analysis. Furthermore, the TLR2 protein was not detectable by Western blot analysis of cells from mice homozygous for the TLR2 knockout allele as compared with cells from TLR2-positive wild-type controls.
Reagents and mAbs
The mAbs specific for CD11c (biotin-labeled: 09702D, PE-labeled: 09705B), streptavidin-CyChrome (13038A) as well as PE-labeled MHC class II (06355A), CD40 (09665B), CD80 (09605B), and CD86 (09275B) and corresponding isotype controls were purchased from BD PharMingen (Hamburg, Germany). The mAbs binding a genus-specific epitope of the chlamydial endotoxin (ACI-P500/DS-281099 and ACI-FITC/DS-250298) came from Progen Biotechnik (Heidelberg, Germany). The unlabeled chlamydia-specific Ab was conjugated with an Alexa 546 Protein labeling kit (A-10237) according to the manufacturer’s protocol (Molecular Probes, Leiden, The Netherlands). Saponin (S7900), cycloheximide (C7698), and LPS of Escherichia coli (L2637) were purchased from Sigma (Deisenhofen, Germany), and Urografin (3,5-diacetoamido-2,4,6-triisobenzoic acid) was obtained from Schering (Berlin, Germany).
Culture and multiplication of C. pneumoniae
C. pneumoniae CM-1 (VR-1360; American Type Culture Collection, Manassas, VA) were multiplied according to Maass et al. (41). Chlamydial elementary bodies were centrifuged (2000 × g, 60 min, 35°C) on confluent monolayers of HEp2 cells in the presence of cycloheximide (1 μg/ml). After 72 h of culture, the harvested cells were disrupted with glass beads, and chlamydial elementary bodies were purified in a sucrose urografin gradient (bottom layer, 50% w/v sucrose solution; top layer, 30% v/v urografin in 30 mM Tris-HCl buffer, pH 7.4) at 9000 × g and 4°C for 60 min. After one wash step with 0.2 μm filtered PBS (pH 7.4), purified elementary bodies were stored in SPG buffer (0.22 M sucrose, 8.6 mM Na2HPO4, 3.8 mM KH2PO4, 5 mM glutamic acid, 0.2 μm filtered, pH 7.4) at −70°C until use. To quantify the number of elementary bodies, HEp2 cells were infected and stained with the chlamydia-specific Ab. The number of inclusion-forming units (IFU) was counted as determined by fluorescence microscopy (Carl Zeiss Jena, Göttingen, Germany) 48 h after infection. For control, noninfected HEp2 cells were treated in the same way. Contamination with mycoplasma was excluded regularly by Mycoplasma-PCR using specific primers (MWG Biotech, Martinsried, Germany).
Isolation and differentiation of BMDDC
BMDDC were generated according to Lutz et al. (42) with slight modifications. Briefly, femora and tibiae of mice were rinsed with cell culture medium applied through a 26-gauge syringe. Bone marrow cells were cultured in bacterial petri dishes at a density of 4 × 106 cells/dish in the presence of 200 U/ml GM-CSF. The medium used was very low endotoxin RPMI 1640 (Biochrom KG, Berlin, Germany) supplemented with 10% FBS (Biochrom KG) and 2-ME (50 μM; Life Technologies, Karlsruhe, Germany). After 3 days of culture, fresh medium supplemented with GM-CSF (200 U/ml) was added. On day 6 of culture, nonadherent cells were transferred to six-well cell culture dishes at a density of 0.75 × 106 cells and 2 ml of fresh medium with GM-CSF (200 U/ml) per well was added. BMDDC were exposed to C. pneumoniae (5 IFU/cell unless otherwise indicated) on the 7th day of culture in the absence of antibiotics and cycloheximide, and without centrifugation.
Detection of cytokines
The cytokines IL-12p40 and TNF-α were determined in the culture supernatant of BMDDC by commercially available ELISAs (dual set for IL-12p40 and TNF-α; R&D Systems, Wiesbaden-Nordenstadt, Germany). The assays were performed as described by the manufacturer. Cytokine determinations were performed at least in duplicates.
FACS analysis
Dendritic cells were double stained (30 min, 4°C) after 48 h of exposure to C. pneumoniae with mAb directed against CD11c (2 μg/ml) and MHC class II (1 μg/ml), CD11c and CD40 (2 μg/ml), CD11c and CD80 (2 μg/ml), or CD11c and CD86 (2 μg/ml). After three wash steps the cells were fixed with 1% paraformaldehyde and analyzed with a FACSCalibur instrument (BD Biosciences, San Jose, CA).
Confocal microscopy
To analyze chlamydial development inside dendritic or HEp2 cells, the cells were cultured on glass platelets and exposed to C. pneumoniae for 48 h. The glass platelets were removed and, in the case of BMDDC, stained with a FITC-labeled CD11c-specific Ab (30 min, 4°C). After three wash steps the cells were fixed with paraformaldehyde (1% w/v, 30 min, 4°C), which was followed by another wash cycle. Thereafter, cells were permeabilized with saponin (0.5% w/v, 30 min, 4°C) and stained with the Alexa 546-labeled chlamydia-specific mAb. The cells were washed again and analyzed by confocal microscopy (LSM510; Carl Zeiss Jena).
Detection of NF-κB via EMSA
Nuclear protein extracts were prepared from BMDDC using a modified version of a published protocol (43). Briefly, BMDDC were exposed to C. pneumoniae for the time period indicated and harvested subsequently by gentle pipetting. The suspension (∼3 × 106 cells) was washed once with 1 ml of ice-cold PBS. Cells were pelleted (2 min, 2000 × g), lysed in 50 μl of ice-cold buffer A (10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 5 mM DTT, 300 mM sucrose, 0.1% Nonidet P-40, 7.5 μg/ml pepstatin, antipain, chymostatin, leupeptin, and aprotinin, and 5 mM PMSF), and left on ice for 5 min. The nuclei were pelleted by pulse centrifugation, resuspended in 100 μl buffer A, pulse centrifuged again, and the supernatant was discarded. Fifty microliters of ice-cold buffer B (20 mM HEPES (pH 7.9), 20% glycerol, 100 mM KCl, 100 mM NaCl, 5 mM PMSF, 7.5 μg/ml pepstatin, antipain, chymostatin, leupeptin, and aprotinin) was added, and nuclei were sonicated for 10 s, snap-frozen, and stored at −70°C until use. Protein concentration was determined using the Bradford method (Bio-Rad, Munich, Germany).
The prototypic Ig κ-chain oligonucleotide was used as a probe and labeled by annealing of complementary primer extension with the Klenow fragment of DNA polymerase I (Roche Diagnostics, Mannheim, Germany) in the presence of [α-32P]dCTP (3000 Ci/mmol; NEN, Brussels, Belgium) and deoxynucleoside triphosphates (Roche Diagnostics). Nuclear extracts (4 μg protein) were incubated with radiolabeled DNA probes (∼10 ng; 105 cpm) for 30 min at room temperature in 20 μl of binding buffer (20 mM HEPES (pH 7.9), 50 mM KCl, 1 mM DTT, 0.5 mM EDTA, 10% glycerol, 1 mg/ml BSA, 0.2% Nonidet P-40, 50 ng/μl poly(dI-dC)). Samples were run in 0.25× TBE buffer (22.3 mM Tris, 22.3 mM boric acid, 0.5 mM EDTA pH 8.0) on nondenaturing 4% polyacrylamide gels at 125 V. As a control the binding Sp-1 activity was analyzed in the same nuclear extracts by EMSA using specific consensus oligonucleotides (Promega, Heidelberg, Germany) that were labeled with [γ-32P]ATP (5000 Ci/mmol; NEN) and T4 polynucleotide kinase (Roche Diagnostics). Gels were dried and visualized by autoradiography.
Luciferase assay
Luciferase assays on TLR2-transfected human embryonic kidney (HEK)293 cells were performed as described (44). The expression vector for murine TLR2 was provided by H. Heine (Research Center, Borstel, Germany) and D. Golenbock (Evans Biomedical Research Center, Boston, MA; Ref. 45). Briefly, 3 × 105 HEK293 cells were plated in 3.5-cm dishes and transfected the following day with 0.1 μg of endothelial cell-leukocyte adhesion molecule-1 luciferase reporter construct, 1 μg of Rous sarcoma virus-β-galactosidase plasmid for normalization, and 0.2 μg of an expression plasmid encoding murine TLR2. Twenty-four hours after transfection, cells were stimulated with purified C. pneumoniae. Six to 8 h later, the cells were lysed for measurement of luciferase activity using reagents from Promega (Madison, WI). The results were normalized to β-galactosidase activity.
Results
Stimulation of BMDDC by C. pneumoniae
BALB/c BMDDC, generated by a 7-day culture in the presence of GM-CSF, were exposed to C. pneumoniae, and 2 days later their morphology was analyzed by phase contrast microscopy. BMDDC reacted with long cytoplasmic extensions upon exposure to C. pneumoniae (data not shown). Fluorescence microscopy revealed that at this point in time the chlamydial material visible within BMDDC did not resemble inclusions generated in the permissive epithelial carcinoma cell line HEp2 but was similar in appearance to inclusions found in macrophages (Fig. 1) (4). The expression of MHC class II, CD40, CD80, and CD86 was up-regulated clearly after a 2-day exposure to C. pneumoniae as shown by the FACS analysis depicted in Fig. 2. Similar results were obtained with dendritic cells from C57BL/6 mice (data not shown). At the same point in time, the ability of C. pneumoniae-exposed BALB/c BMDDC to take up FITC-labeled dextran particles was impaired indicating that BMDDC were matured (Fig. 3). Thus, BMDDC mice were stimulated to mature upon contact with C. pneumoniae.

Appearance of C. pneumoniae in BMDDC and HEp2 cells. BMDDC and HEp2 cells were analyzed by confocal microscopy after 2 days of contact with C. pneumoniae (10 IFU/cell). Cells were treated with paraformaldehyde (1%) and saponin (0.5%) and stained with the Alexa 546-labeled Ab specific for chlamydial endotoxin to demonstrate the intracellular presence of C. pneumoniae. To show that C. pneumoniae was indeed observed in dendritic cells, BMDDC were first stained with a FITC-labeled Ab specific for CD11c. Subsequently, the cells were treated with paraformaldehyde and saponin and stained for intracellular C. pneumoniae with a Alexa 546-labeled Ab specific for chlamydial endotoxin. The experiment was performed three times with equal results.
Appearance of C. pneumoniae in BMDDC and HEp2 cells. BMDDC and HEp2 cells were analyzed by confocal microscopy after 2 days of contact with C. pneumoniae (10 IFU/cell). Cells were treated with paraformaldehyde (1%) and saponin (0.5%) and stained with the Alexa 546-labeled Ab specific for chlamydial endotoxin to demonstrate the intracellular presence of C. pneumoniae. To show that C. pneumoniae was indeed observed in dendritic cells, BMDDC were first stained with a FITC-labeled Ab specific for CD11c. Subsequently, the cells were treated with paraformaldehyde and saponin and stained for intracellular C. pneumoniae with a Alexa 546-labeled Ab specific for chlamydial endotoxin. The experiment was performed three times with equal results.
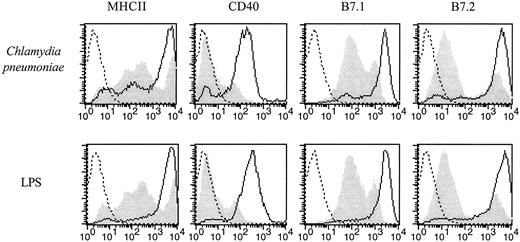
Up-regulation of MHC class II, CD40, CD80, and CD86 by dendritic cells derived from BALB/c mice upon contact with C. pneumoniae. BMDDC derived from BALB/c mice were exposed to C. pneumoniae (5 IFU/cell) or stimulated with endotoxin (50 ng/ml) for 2 days. The cells were double stained with a strepatavidin-CyChrome-labeled, CD11c-specific mAb and a PE-labeled anti-MHC class II mAb, a PE-labeled anti-CD40 mAb, a PE-labeled anti-CD80 mAb, or a PE-labeled anti-CD86 mAb as indicated in the graphs. The cells depicted in the graphs were gated on CD11c. Dead cells were excluded based on forward and side scatter characteristics. Dotted lines represent isotype controls, the gray shadowed area represents dendritic cells exposed to a preparation of uninfected HEp2 cells (upper panels) or medium alone (lower panels), and the solid line shows dendritic cells exposed to C. pneumoniae. The experiment was performed three times with equal results.
Up-regulation of MHC class II, CD40, CD80, and CD86 by dendritic cells derived from BALB/c mice upon contact with C. pneumoniae. BMDDC derived from BALB/c mice were exposed to C. pneumoniae (5 IFU/cell) or stimulated with endotoxin (50 ng/ml) for 2 days. The cells were double stained with a strepatavidin-CyChrome-labeled, CD11c-specific mAb and a PE-labeled anti-MHC class II mAb, a PE-labeled anti-CD40 mAb, a PE-labeled anti-CD80 mAb, or a PE-labeled anti-CD86 mAb as indicated in the graphs. The cells depicted in the graphs were gated on CD11c. Dead cells were excluded based on forward and side scatter characteristics. Dotted lines represent isotype controls, the gray shadowed area represents dendritic cells exposed to a preparation of uninfected HEp2 cells (upper panels) or medium alone (lower panels), and the solid line shows dendritic cells exposed to C. pneumoniae. The experiment was performed three times with equal results.
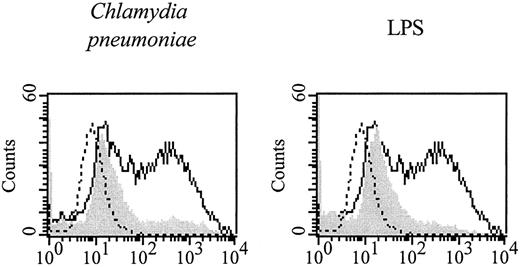
Maturation of dendritic cells upon exposure to C. pneumoniae. BMDDC derived from BALB/c mice were exposed to C. pneumoniae (5 IFU/cell, left graph) or endotoxin (50 ng/ml, right graph) for 2 days. Subsequently, cells were incubated with FITC-labeled dextran (m.w. 40,000, 0.1 mg/ml) for 2 h, and the uptake of dextran was analyzed by FACS (gray shadowed area). BMDDC exposed to a preparation of uninfected HEp2 cells served as controls (solid line). Dotted lines depict cells that were stimulated with a preparation of uninfected HEp2 cells and not exposed to dextran. The experiment was performed three times with equal results.
Maturation of dendritic cells upon exposure to C. pneumoniae. BMDDC derived from BALB/c mice were exposed to C. pneumoniae (5 IFU/cell, left graph) or endotoxin (50 ng/ml, right graph) for 2 days. Subsequently, cells were incubated with FITC-labeled dextran (m.w. 40,000, 0.1 mg/ml) for 2 h, and the uptake of dextran was analyzed by FACS (gray shadowed area). BMDDC exposed to a preparation of uninfected HEp2 cells served as controls (solid line). Dotted lines depict cells that were stimulated with a preparation of uninfected HEp2 cells and not exposed to dextran. The experiment was performed three times with equal results.
BMDDC secrete proinflammatory cytokines upon stimulation with C. pneumoniae
Microbial pattern ligands like endotoxin or CpG DNA stimulate murine BMDDC to secrete proinflammatory cytokines including TNF-α and IL-12 (20). Therefore, we tested whether C. pneumoniae would trigger similar responses from BMDDC. Fig. 4 shows that BMDDC from BALB/c mice secreted large amounts of TNF-α with peak levels ∼4 h after exposure. IL-12p40 was also detected in the culture supernatant and peaked 24 h after exposure (Fig. 4). In contrast, IL-12p70 and IL-10 levels were beyond the detection limits of the ELISA system (data not shown).

Kinetics of TNF-α and IL12 p40 production by BALB/c dendritic cells upon contact with C. penumoniae. Dendritic cells from BALB/c mice were exposed to C. pneumoniae for the indicated periods of time. Thereafter, the amount of TNF-α and IL-12p40 of the culture supernatant was analyzed by ELISA. Cells exposed to a preparation of uninfected HEp2 cells served as control (HEp2). The experiment was performed twice with equal results.
Kinetics of TNF-α and IL12 p40 production by BALB/c dendritic cells upon contact with C. penumoniae. Dendritic cells from BALB/c mice were exposed to C. pneumoniae for the indicated periods of time. Thereafter, the amount of TNF-α and IL-12p40 of the culture supernatant was analyzed by ELISA. Cells exposed to a preparation of uninfected HEp2 cells served as control (HEp2). The experiment was performed twice with equal results.
Role of TLR
BMDDC derived from C3H/HeN mice express TLR2 and TLR4 as analyzed by RT-PCR (data not shown). Therefore, we analyzed the requirement of these TLRs for the recognition of C. pneumoniae by BMDDC derived from TLR4-mutant C3H/HeJ mice and TLR2-deficient mice. BMDDC from TLR4-mutant C3H/HeJ mice as well as BMDDC from C3H/HeN control mice up-regulated the expression of MHC class II, CD40, CD80, and CD86 upon exposure to C. pneumoniae, suggesting that a functional TLR4 was not required (data not shown). Furthermore, the analysis of TNF-α secretion after stimulation with titrated amounts of C. pneumoniae showed that BMDDC from TLR4-mutant C3H/HeJ mice secreted equal amounts of TNF-α as did BMDDC from normal C3H/HeN mice (Fig. 5,A). Control stimulation with endotoxin revealed the expected result that BMDDC from endotoxin-hyporesponsive C3H/HeJ mice failed to secrete TNF-α, yet BMDDC from C3H/HeN, TLR2-deficient, and wild-type control mice responded well (Fig. 5,A). In contrast, the release of TNF-α by TLR2- deficient BMDDC was severely impaired upon exposure to C. pneumoniae (Fig. 5,A). In addition, HEK293 cells transfected with a NF-κB-luciferase reporter construct responded to C. pneumoniae provided they were cotransfected with a murine TLR2 expression plasmid (Fig. 6). Similarly, peritoneal washout macrophages from TLR2-deficient or TLR4-mutant mice displayed the same TLR dependence of TNF-α secretion upon exposure to C. pneumoniae (data not shown). Thus, this phenotype is not confined to BMDDC. In contrast to these observations, production of IL-12p40 by C. pneumoniae-stimulated BMDDC appeared to be dependent on both TLR2 and TLR4. As shown in Fig. 5 B, the secretion of IL-12p40 by TLR4-mutant or TLR2-deficient dendritic cells reached only ∼50% of the level of the respective control BMDDC. Taken together, these data suggest that both TLR2 and TLR4 are involved in the recognition of C. pneumoniae by BMDDC. However, their respective importance depends on the BMDDC response analyzed. Thus, TLR2 is essential for TNF-α secretion, whereas for optimal IL-12p40 secretion both TLR2 and TLR4 are required.

TNF-α and IL-12p40 response of BMDDC derived from TLR2-deficient and TLR4-mutant mice upon stimulation with C. pneumoniae. BMDDC derived from TLR4-mutant C3H/HeJ and normal C3H/HeN control mice as well as TLR2-deficient and wild-type control mice were exposed on day 7 of GM-CSF-induced differentiation to titrated amounts of C. pneumoniae for 2 days. In parallel the cells were stimulated with endotoxin (50 ng/ml) for 2 days. A, Amount of TNF-α; B, amount of IL-12p40 produced. Upper graphs in A and B, Response of C3H/HeJ (□) and C3H/HeN mice (▪); lower graphs in A and B, response of TLR2-deficient (□) and wild-type control mice (▪). Negative controls include exposure of BMDDC to a preparation of uninfected HEp2 cells (HEp2) or exposure to medium alone (medium). The experiment was performed three times with equal results.
TNF-α and IL-12p40 response of BMDDC derived from TLR2-deficient and TLR4-mutant mice upon stimulation with C. pneumoniae. BMDDC derived from TLR4-mutant C3H/HeJ and normal C3H/HeN control mice as well as TLR2-deficient and wild-type control mice were exposed on day 7 of GM-CSF-induced differentiation to titrated amounts of C. pneumoniae for 2 days. In parallel the cells were stimulated with endotoxin (50 ng/ml) for 2 days. A, Amount of TNF-α; B, amount of IL-12p40 produced. Upper graphs in A and B, Response of C3H/HeJ (□) and C3H/HeN mice (▪); lower graphs in A and B, response of TLR2-deficient (□) and wild-type control mice (▪). Negative controls include exposure of BMDDC to a preparation of uninfected HEp2 cells (HEp2) or exposure to medium alone (medium). The experiment was performed three times with equal results.

Activation of a NF-κB reporter construct upon stimulation with C. pneumoniae depends on TLR2. HEK293 cells were triple transfected with a NF-κB-luciferase reporter construct, a β-galactosidase-containing plasmid, as well as TLR2; control cells were also triple transfected, with the difference that instead of TLR2 only the empty plasmid was used. Twenty-four hours later the cells were stimulated with C. pneumoniae (20 IFU/cell) or a control preparation from uninfected HEp2 cells or were exposed to medium alone. After 48 h the activity of luciferase was quantified and normalized to the activity of β-galactosidase. The experiment was performed twice with equal results.
Activation of a NF-κB reporter construct upon stimulation with C. pneumoniae depends on TLR2. HEK293 cells were triple transfected with a NF-κB-luciferase reporter construct, a β-galactosidase-containing plasmid, as well as TLR2; control cells were also triple transfected, with the difference that instead of TLR2 only the empty plasmid was used. Twenty-four hours later the cells were stimulated with C. pneumoniae (20 IFU/cell) or a control preparation from uninfected HEp2 cells or were exposed to medium alone. After 48 h the activity of luciferase was quantified and normalized to the activity of β-galactosidase. The experiment was performed twice with equal results.
C. pneumoniae-induced nuclear translocation of NF-κB in BMDDC is dependent on TLR2
The transcription factor NF-κB participates in the transcriptional regulation of a variety of immune response genes like MHC class II genes and cytokine genes (46). Because MHC class II molecules are up-regulated (data not shown) and TNF-α is secreted by BMDDC from C3H/HeJ mice exposed to C. pneumoniae, we expected that NF-κB is also activated in BMDDC of TLR4-mutant mice. Indeed, as shown in Fig. 7 exposure of BMDDC from C3H/HeJ and C3H/HeN mice to C. pneumoniae for 1, 2, and 5 h or TNF-α induced nuclear translocation of NF-κB. In contrast, stimulation with endotoxin translocated NF-κB, as expected, only in dendritic cells from C3H/HeN mice (Fig. 7). In contrast, the extent of nuclear translocation of NF-κB in dendritic cells from TLR2-deficient mice was attenuated strongly upon contact with C. pneumoniae in comparison to dendritic cells from wild-type control mice at all time points analyzed (Fig. 7). However, both types of BMDDC responded equally to stimulation with endotoxin (Fig. 7). Taken together, C. pneumoniae-derived pattern ligands are recognized via TLR2, which in turn activates NF-κB, presumably via the IL-1R/TLR signal pathway involving MyD88 and TNFR-associated factor 6 (44, 47, 48).

C. pneumoniae-induced nuclear translocation of NF-κB is independent of TLR4 yet depends on TLR2. BMDDC from TLR4-mutant C3H/HeJ and C3H/HeN control mice (upper graphs) were exposed on day 7 of GM-CSF-induced maturation to C. pneumoniae (5 IFU/cell) for the time periods indicated, endotoxin (50 ng/ml) for 2 h, or TNF-α (5 ng/ml) for 2 h. Unstimulated cells (no stim.) and cells exposed to a control preparation from uninfected HEp2 cells (HEp2 lys.) served as negative controls. The same experiment described above was performed with BMDDC from TLR2-deficient and wild-type control mice (lower graphs). Nuclear extracts were prepared, and an EMSA was performed as described in Materials and Methods for detection of nuclear translocation and DNA binding activity of NF-κB. Sp1 served as loading control. Specificity of the radioactive labeled NF-κB or Sp1 probe was shown by competition with the unlabeled corresponding oligonucleotide (LPS+comp., upper and lower graphs). The experiment was performed three times with equal results.
C. pneumoniae-induced nuclear translocation of NF-κB is independent of TLR4 yet depends on TLR2. BMDDC from TLR4-mutant C3H/HeJ and C3H/HeN control mice (upper graphs) were exposed on day 7 of GM-CSF-induced maturation to C. pneumoniae (5 IFU/cell) for the time periods indicated, endotoxin (50 ng/ml) for 2 h, or TNF-α (5 ng/ml) for 2 h. Unstimulated cells (no stim.) and cells exposed to a control preparation from uninfected HEp2 cells (HEp2 lys.) served as negative controls. The same experiment described above was performed with BMDDC from TLR2-deficient and wild-type control mice (lower graphs). Nuclear extracts were prepared, and an EMSA was performed as described in Materials and Methods for detection of nuclear translocation and DNA binding activity of NF-κB. Sp1 served as loading control. Specificity of the radioactive labeled NF-κB or Sp1 probe was shown by competition with the unlabeled corresponding oligonucleotide (LPS+comp., upper and lower graphs). The experiment was performed three times with equal results.
Discussion
This study shows that BMDDC are stimulated to mature upon exposure to C. pneumoniae. Expression of MHC class II and costimulatory molecules like CD40, CD80, and CD86 was up-regulated, NF-κB was activated, and proinflammatory cytokines were secreted. In contrast to endotoxin-induced BMDDC stimulation, the responses depend mainly on TLR2 and only partially on TLR4.
The chlamydial material observed in BMDDC by fluorescence microscopy resembles structures seen in macrophages, i.e., small, not completely developed inclusions (4). Ultrastructural analysis of these structures in macrophages by electron microscopy revealed small inclusions that contain only a few chlamydial particles (4). Furthermore, chlamydia within these small inclusions seem to persist and are metabolically active (4). Similar aberrant forms develop in IFN-γ-treated HeLa cells infected with C. trachomatis (49). Upon removal of IFN-γ, typical inclusions develop together with a new progeny of infectious elementary bodies (50). The replication of C. pneumoniae in HEp2 cells is also inhibited upon treatment of cells with IFN-γ, TNF-α, or lymphotoxin (51, 52), and in one study IFN-γ treatment appeared to induce aberrant and persistent forms of C. pneumoniae in HEp2 cells (53). As shown here (Fig. 4), BMDDC secrete rapidly large amounts of TNF-α upon contact with C. pneumoniae. We speculate that TNF-α causes restriction of chlamydial development, which may result in persistence of C. pneumoniae. If so, BMDDC from TNFR p55-deficient mice may not restrict chlamydial development. In vivo mice lacking the IFN-γ receptor or iNOS are highly susceptible to infection with C. pneumoniae (16). We have not yet analyzed whether BMDDC produce NO upon exposure to C. pneumoniae and whether this mechanism is responsible for the restricted development in BMDDC. However, in mice infected with Leishmania major a substantial part of L. major foci were associated with iNOS-positive macrophages or dendritic cells, and pharmacological inhibition of iNOS caused an increase of the parasite burden (54).
Pathogen-derived pattern ligands like endotoxin and CpG DNA stimulate dendritic cells to increase the expression of MHC class II, CD40, CD80, and CD86 and to secrete cytokines like TNF-α or IL-12p40 (20). It is at present unclear which component of C. pneumoniae is responsible for the activation of BMDDC. Because chlamydial lipid A contains only five instead of six fatty acids with a chain length of 14–20 C atoms the stimulatory capacity of chlamydial endotoxin has been reported as weak (10, 11, 12). To explore whether chlamydial endotoxin participates in the activation of BMDDC we analyzed the response of endotoxin-hyporeactive C3H/HeJ BMDDC. Here we describe that NF-κB activation and translocation, TNF-α secretion, and up-regulation of costimulatory molecules of C3H/HeJ BMDDC was as strong as in control C3H/HeN BMDDC (Figs. 5 and 7, and data not shown). Furthermore, polymyxin B failed to block stimulation of dendritic cells by C. pneumoniae in terms of TNF-α and IL-12p40 secretion (data not shown). These findings suggest that chlamydial endotoxin is unable to stimulate dendritic cells via TLR4 if they are exposed to the intact microorganism. Whether chlamydial endotoxin triggers BMDDC via TLR2 or whether a yet undefined C. pneumoniae-derived pattern ligand signals via TLR2 needs to be investigated.
In terms of the responses analyzed here, TLR2 appears more important than TLR4 for C. pneumoniae-induced activation of BMDDC. Translocation of NF-κB as well as the release of cytokines measured in this study were reduced substantially in BMDDC derived from TLR2-deficient mice (Figs. 5 and 7). In addition, activation of an NF-κB luciferase construct in HEK293 cells was dependent on the presence of TLR2 (Fig. 6). These observations do not rule out the possibility that TLRs other than TLR2 and 4 are involved in C. pneumoniae-induced responses of BMDDC not analyzed here. Also, other cell types may use a different set of TLRs to recognize C. pneumoniae. Accordingly, it was recently shown that TLR2 and TLR6 cooperate in macrophage activation by Gram-positive bacteria (34). Thus, a combination of TLR2 and other TLRs could also play a role in C. pneumoniae-induced stimulation of BMDDC. The question remains which chlamydial compound is responsible for the TLR2-mediated activation of BMDDC. Peptidoglycans from Gram-positive bacteria were found to trigger innate immune cells in a TLR2-dependent fashion (23). As already mentioned, this macromolecule could hardly be detected biochemically in Chlamydia (7, 8, 9). In contrast, Chlamydia are sensitive to penicillin G known to interfere with the synthesis of peptidoglycan (55). In light of these findings, it appears unlikely that chlamydial peptidoglycans play a major role in the activation of cells of the innate immune system. Because TLR2 is not involved in CpG DNA-induced cellular activation (31) and the frequency of CpG motifs in the genome of C. pneumoniae is low compared with other bacteria (our unpublished observation), chlamydial CpG DNA is also an unlikely candidate. Despite these considerations the potency of chlamydial DNA to stimulate innate immune cells can now be tested using TLR9-deficient mice because this TLR was shown to mediate activation of innate immune cells by bacterial CpG DNA (24). In search of the stimulatory chlamydial component, we consider one of the chlamydial heat shock proteins (hsp) as a possible candidate. In particular, chlamydial hsp60 was described to activate macrophages to secrete cytokines like TNF-α and to regulate expression of matrix metalloproteinases (56). Indeed, using purified hsp60 from C. pneumoniae we are now able to show that this protein stimulates potently innate immune cells (C. Prazeres da Costa, C. Kirschning, D. Busch, S. Prebeck, S. Dürr, H. Wagner, and T. Miethke, manuscript in preparation).
The dominant role of TLR2 vs TLR4 in the recognition process of C. pneumoniae by BMDDC was surprising because we expected initially that for recognition of a Gram-negative bacterium like C. pneumoniae TLR4 would be more relevant. TLR2 was demonstrated to be involved in the recognition of components of the Gram-positive cell wall (23, 33) as well as whole bacteria like S. aureus, Listeria monocytogenes, Borrelia burgdorferi, and Mycobacterium avium (33, 57). Furthermore, a dominant-negative mutant of TLR2 inhibited TNF-α secretion in RAW-TT10 macrophages upon stimulation with S. aureus but not with S. minnesota (30). In contrast, TLR4-mutant and -deficient mice show convincingly that TLR4 is involved in the recognition of endotoxin and thus Gram-negative bacteria (23, 28, 58, 59). However, it has been shown that for recognition of a number of Gram-negative endotoxins, TLRs other than TLR4 may be involved additionally (30, 40). Also, both TLR2 and TLR4 appear to be responsible for recognition of Mycobacterium tuberculosis (60). Thus for recognition of diverse components of whole bacteria several TLRs may be needed, and different cellular responses may rely on different TLRs. To our knowledge, this is the first example that for recognition of a whole Gram-negative bacterium by innate immune cells TLR2 appears to be central whereas TLR4 plays only a minor role. Obviously, to study the relative importance of a given TLR for the recognition process of bacteria by immune cells a number of cellular responses have to be analyzed. Thus, one important aspect of this study is that different cellular responses induced by bacteria, as exemplified here with TNF-α and IL-12p40 secretion, may require different TLRs, which may be stimulated by different pathogen-associated pattern ligands.
Does TLR2 play an important role for recognition of C. pneumoniae in vivo? This important question is presently our focus of interest. Until now the best evidence that this is the case came from i.p. injection of C. pneumoniae and restimulation of peritoneal macrophages with endotoxin in vitro. In wild-type mice, stimulation of peritoneal macrophages with C. pneumoniae in vivo induced a hyporesponsiveness of macrophages toward a stimulation with endotoxin in vitro. This phenomenon could not be observed in TLR2-deficient mice (data not shown), indicating that macrophages recognized C. pneumoniae in vivo via TLR2.
Another issue raised by our findings is the function of C. pneumoniae-stimulated BMDDC. Initially, we expected that C. pneumoniae might decrease the expression of MHC class II molecules on the cell surface of BMDDC because this would impede presentation of chlamydial proteins to Ag-specific T cells and would thus generate an immune escape mechanism. Indeed, it has been shown that infection of epithelial cells with C. trachomatis prevents the IFN-γ-induced expression of MHC class II molecules (61). In contrast to our expectation, BMDDC increased the expression of MHC class II molecules as well as costimulatory molecules upon exposure to C. pneumoniae. Therefore, one would predict that C. pneumoniae-specific T cells are activated effectively by BMDDC to limit the spread of C. pneumoniae in vivo. Indeed, dendritic cells pulsed with killed C. trachomatis and injected into mice provide protection against subsequent infection (62). In contrast C. pneumoniae-exposed and -stimulated dendritic cells could also initiate autoimmune responses due to presentation of cross-reactive epitopes. As shown by Penninger et al. (63) injection of chlamydial peptides derived from the 60-kDa cysteine-rich outer membrane protein induced myocarditis due to a cross-reactive epitope with the α myosin heavy chain molecule. Because in that study only peptides were injected it is unclear whether a systemic infection of experimental animals with C. pneumoniae or C. trachomatis would result in autoimmune disease. Clearly, further experimentation is needed to answer these questions.
Acknowledgements
We appreciate the invaluable assistance of Tamara Eisele. We thank Dr. Irene Seegmüller for helping us in RT-PCR analysis of TLR2 and TLR4 expression.
Footnotes
Abbreviations used in this paper: iNOS, inducible NO synthase; TLR, Toll-like receptor; BMDDC, murine dendritic cells derived from bone marrow; IFU, inclusion-forming units; hsp, heat shock protein; HEK, human embryonic kidney.
