

Abstract
Elevated levels of PGE2 have been reported in synovial fluid and cartilage from patients with osteoarthritis (OA). However, the functions of PGE2 in cartilage metabolism have not previously been studied in detail. To do so, we cultured cartilage explants, obtained from patients undergoing knee replacement surgery for advanced OA, with PGE2 (0.1–10 μM). PGE2 inhibited proteoglycan synthesis in a dose-dependent manner (maximum 25% inhibition (p < 0.01)). PGE2 also induced collagen degradation, in a manner inhibitable by the matrix metalloproteinase (MMP) inhibitor ilomastat. PGE2 inhibited spontaneous MMP-1, but augmented MMP-13 secretion by OA cartilage explant cultures. PCR analysis of OA chondrocytes treated with PGE2 with or without IL-1 revealed that IL-1-induced MMP-13 expression was augmented by PGE2 and significantly inhibited by the cycolooygenase 2 selective inhibitor celecoxib. Conversely, MMP-1 expression was inhibited by PGE2, while celecoxib enhanced both spontaneous and IL-1-induced expression. IL-1 induction of aggrecanase 5 (ADAMTS-5), but not ADAMTS-4, was also enhanced by PGE2 (10 μM) and reversed by celecoxib (2 μM). Quantitative PCR screening of nondiseased and end-stage human knee OA articular cartilage specimens revealed that the PGE2 receptor EP4 was up-regulated in OA cartilage. Moreover, blocking the EP4 receptor (EP4 antagonist, AH23848) mimicked celecoxib by inhibiting MMP-13, ADAMST-5 expression, and proteoglycan degradation. These results suggest that PGE2 inhibits proteoglycan synthesis and stimulates matrix degradation in OA chondrocytes via the EP4 receptor. Targeting EP4, rather than cyclooxygenase 2, could represent a future strategy for OA disease modification.
Osteoarthritis (OA)3 is the most common disorder affecting diarthrodial joints and has been associated with age-related loss of homeostatic balance between degeneration and repair mechanisms. OA is often a consequence of abnormal mechanical forces that lead to altered metabolism in chondrocytes, the single cellular component in articular cartilage. Although OA is generally classified as a noninflammatory disease, prominent proinflammatory mediators, including cytokines, PGs, and reactive oxygen species are believed to play pivotal roles in the disease pathogenesis (1, 2, 3, 4, 5, 6). It is increasingly appreciated that chondrocytes, like osteocytes in bone, function as mechanosensors by which local mechanical forces influence cellular metabolism. Abnormal loading applied to chondrocytes in vitro has been shown to induce proteins associated with the catabolic phenotype characteristic of OA (e.g., matrix metalloproteinases (MMPs), inducible NO synthase, cyclooxygenase 2 (COX-2), microsomal prostaglandin E synthase (mPGES), and IL-1), thus providing a pathophysiologic link between the altered mechanical forces and biochemical mediators that characterize OA (7, 8, 9, 10).
We have previously shown that chondrocytes from human OA cartilage explants express COX-2 and spontaneously produce PGE2 (11). Gene expression analysis of both intact and damaged cartilage obtained from OA patients has shown a concomitant increase in the expression of microsomal PG synthase, a terminal synthase required for COX-2-derived PGE2 production in diseased, but not normal cartilage (12). The coordinate expression of COX-2 and mPGES can be further up-regulated in chondrocytes by the proinflammatory cytokines IL-1β and TNF-α (13, 14, 15). Recently, Beier’s group (16) confirmed up-regulation of COX-2 in vivo in a surgically induced model of OA. These observations suggest that PGs may contribute to disease pathogenesis in OA in a manner that is distinct from their capacity to promote vasodilation and pain, characteristic of classical inflammation in vascularized tissues. However, the role of prostanoid overproduction on anabolic and catabolic functions of cartilage remains incompletely elucidated.
We have examined the effects of PGE2 on OA cartilage and have begun to characterize the signaling pathway responsible for those effects. PGE2 is known to bind to four distinct cell surface receptors (EP1, EP2, EP3, and EP4), each of which is a “serpentine seven” G protein-coupled receptor; ligation of individual receptors mediates distinct intracellular signaling pathways that may contribute to the pleiotropic effects of PGs in different tissues (17, 18). In the current study, we report that the predominant effects of PGE2 in OA chondrocytes are catabolic and show for the first time that these effects are mediated via EP4 receptor signaling.
Materials and Methods
Reagents
Affymetrix gene chips (U95Av2A) were obtained from Affymetrix. Matrix metalloproteinases proMMP-1 and proMMP-13 ELISA kits were purchased from R&D Systems. EP4 antagonist (AH23848), EP2 antagonist (AH6809), and PGE2 were obtained from Sigma-Aldrich. Cell culture media were purchased from Invitrogen. IL-1β and MMP inhibitor (Ilomastat) were from PeproTech and Chemicon International, respectively. Reagents for real-time PCR were from Applied Biosystems. Other reagents are as described below.
Procurement of human cartilage
Cartilage slices were obtained from the knees of patients (age 50–70 years) with advanced OA and undergoing knee replacement surgery. Nonarthritic knee cartilages were obtained from autopsy patients within 24 h (National Diabetes Research Interchange, Philadelphia, PA). The use of discarded human cartilage was approved by appropriate institutional review boards.
Isolation of human chondrocytes from cartilage
Cartilage obtained at surgery was cut into small pieces and digested with collagenase (0.1%) for 12–16 h in Ham’s F-12 medium. This cell suspension was used to establish cell cultures and maintained at 37°C in a humidified atmosphere of 95% air/5% CO2. Chondrocytes were maintained as monolayer cultures for not more than 3–4 days before use to avoid loss of the chondrocyte phenotype (19).
Explant culture of OA cartilage
Briefly, articular cartilage from patients undergoing knee replacement surgery was obtained. The cartilage was cut into 3-mm discs and mixed well and were placed in a 24-well plate in 2.0 ml of Ham’s F-12 medium supplemented with 0.2% endotoxin-free human albumin, 10 mM HEPES (pH 7.4), and antibiotics. Explants were treated with various modulators for 24–72 h and culture supernatants were harvested and analyzed for either NO measured as nitrite released (Greiss method) or secreted PGE2 (RIA; Sigma-Aldrich) (19).
Determination of PGE2
PGE2 was determined in the culture supernatant using a RIA, as reported previously, with a detection limit of 1.0 pg/ml. The values were expressed as nanogram per milliliter of PGE2 released per gram wet weight of cartilage (11).
Proteoglycan synthesis
OA cartilage explant cultures were incubated in Ham’s F-12 culture medium supplemented with l-glutamine (2 mM), gentamicin (50 mg/ml), amphotericin B (0.25 mg/ml), and human albumin (0.2%) in the presence and absence of various modulators for 6 days. At the end of the experiment, 10 μCi/ml sodium sulfate (Na235SO4) was added for 4 h at 37 °C in a 5% CO2 atmosphere and explants were washed five times with 0.15 M NaCl and solubilized in 0.5 ml of Soluene 350 (Packard) in scintillation counting tubes. After addition of 4.5 ml of liquid scintillation counting fluid (Hionic Fluor; Packard Instrument), the 35S-labeled PG content was measured using a Beckman LS 7000 counter (19).
Type II collagen and aggrecan degradation assay
Degradation products of type II collagen and aggrecan were measured using C1,2C and CS846 epitope-based assay kits, respectively, according to the instructions of the manufacturer (IBEX). Values were normalized using wet weight of cartilage, and expressed as fragments released per gram of cartilage.
RNA extraction from articular chondrocytes
For analysis of gene expression following stimulation with IL-1/PGE2, RNA was extracted from monolayer cultures of chondrocytes using TRIzol (Invitrogen). Total RNA was precipitated using isopropanol and further purified using micro-RNeasy columns (Qiagen) as recommended by the manufacturer. RNA content was estimated using a spectrophotometer, the 260:280 ratio for purified RNA was ∼2.0. The integrity of RNA was confirmed using 1% formaldehyde agarose gel electrophoresis (19, 20).
RNA isolation from OA cartilage explants
Cartilage discs were milled into fine powder in liquid nitrogen. RNA was extracted in TRI Reagent (MRC Labs) for 4 h on a rocker, and total RNA was precipitated with equal volumes of isopropanol. The RNA pellet was further purified using a Qiagen RNeasy mini kit according to the manufacturer’s RNA clean-up protocol (19, 20).
Quantification of mRNA by real-time PCR
Total RNA (1 μg) was primed using oligo(dT)18 primers and synthesized according to the manufacturer’s directions. TaqMan primer sets were purchased as predesigned oligonucleotides from Applied Biosystems. Real-Time PCR were run on an Applied Biosystems Prism 7300 sequence detection system. mRNA levels were normalized by using G3PDH as a housekeeping gene and relative expression levels of various transcripts were calculated using an approximation method or the 2-Δ CT method (21).
Flow cytometric analysis of EP receptors in chondrocytes
Expression of EP receptor proteins was assessed on fixed, permeabilized human primary chondrocytes. Briefly, chondrocytes were released using trypsin-EDTA, spun at 1000 rpm for 10 min, and washed thoroughly twice with PBS. The cells were fixed and permeabilized using eBioscience buffers as recommended by the manufacturer. Rabbit polyclonal Abs specific for each EP receptor (Cayman Chemical) was used at 2 μg/sample or isotype-matched control Ab and donkey anti-rabbit labeled with PE (Sigma-Aldrich) was used as secondary Ab. The samples were analyzed with Imascan (BD Biosciences).
Statistics
Data are expressed as mean ± SD. Student’s t test was used to analyze significance and differences with p < 0.05 were considered significant.
Results
PGE2 stimulates MMP-13, but inhibits MMP-1 secretion from OA cartilage
We previously demonstrated that PGE2 inhibits MMP-1, but not MMP-13 production by fibroblast-like synoviocytes (23). In this study, we performed studies on OA cartilage explants to determine the effect of IL-1β and PGE2 on the production of both MMPs by human chondrocytes. Relative to untreated control specimens, addition of IL-1β stimulated both proMMP-13 (Fig. 1,A) and proMMP-1 secretion (Fig. 1 B). However, the stimulatory effect of IL-1β on proMMP-1 was much lower compared to proMMP-13. This may be explained by the high levels of MMP-1 release under basal conditions, which, if near maximal, would result in diminished capacity for induction in response to exogenous factors.

PGE2 stimulates MMP-13, but inhibits MMP-1 secretion from OA cartilage. Human OA cartilage was incubated in triplicate with or without PGE2 (0.1–10 μM) in 24-well plates in serum-free medium. Secretion of MMP-13 (A) and MMP-1 (B) were determined 24 h after stimulation in the culture supernatant. IL-1β served as a positive control. The data represents mean ± SD (n = 4). ∗, p < 0.05 and ∗∗, p < 0.01 performed with four individual patient cartilage explants cultures.
PGE2 stimulates MMP-13, but inhibits MMP-1 secretion from OA cartilage. Human OA cartilage was incubated in triplicate with or without PGE2 (0.1–10 μM) in 24-well plates in serum-free medium. Secretion of MMP-13 (A) and MMP-1 (B) were determined 24 h after stimulation in the culture supernatant. IL-1β served as a positive control. The data represents mean ± SD (n = 4). ∗, p < 0.05 and ∗∗, p < 0.01 performed with four individual patient cartilage explants cultures.
Because of its known ability to induce PGE2 via COX-2 activation, we speculated that IL-1β modulation of MMP expression is due to induction of PGE2. Accordingly, PGE2 addition (1–10 μM) to OA cartilage increased proMMP-13 (Fig. 1,A) and inhibited proMMP-1 secretion (Fig. 1 B) in a dose-dependent manner. These effects parallel our previous observations in fibroblast-like synoviocytes, demonstrating disparate effects of proMMP-1 and proMMP-13 expression in response to PGE2.
PGE2 inhibits proteoglycan synthesis in OA cartilage
Addition of PGE2 (0.1–10 μM) to OA explant cultures decreased proteoglycan synthesis measured as 35S incorporation in a dose-dependent manner (Fig. 2,A). Inhibition of proteoglycan synthesis by PGE2 (10 μM) was comparable to that caused by IL-1β, which suggested a possible role for PGE2 generation in mediating the IL-1β effect on proteoglycan synthesis. We consequently used the COX-2-selective inhibitors celecoxib and rofecoxib to study the role of PGE2 on proteoglycan synthesis. Celecoxib and rofecoxib (2 μM) each inhibited spontaneous PGE2 production by >90% (data not shown) and concurrently augmented (p < 0.01) proteoglycan synthesis (Fig. 2 B). Addition of exogenous PGE2 (10 μM) significantly (p < 0.01) reversed the effects of the celecoxib. Taken together, these data indicate a central role for PGE2 in the inhibition of proteoglycan synthesis by IL-1.

A, PGE2 inhibits proteoglycan synthesis in OA cartilage. Human OA cartilage was incubated with PGE2 (0.1–10 μM) for 6 days in serum-free medium, followed by determination of proteoglycan as 35S incorporation. IL-1β was used as positive control. B, Selective COX-2 inhibitors abrogate proteoglycan synthesis by OA cartilage. Human OA cartilage was incubated with PGE2 (10 μM) with or without the COX-2-selective inhibitors celecoxib or rofecoxib (2 μM) for 6 days in serum-free medium. Medium was changed every 48 h. Proteoglycan synthesis was determined as 35S incorporation as described in Materials and Methods. The data represents mean ± SD (n = 5). ∗, p < 0.01 performed with five individual patient cartilage explants cultures.
A, PGE2 inhibits proteoglycan synthesis in OA cartilage. Human OA cartilage was incubated with PGE2 (0.1–10 μM) for 6 days in serum-free medium, followed by determination of proteoglycan as 35S incorporation. IL-1β was used as positive control. B, Selective COX-2 inhibitors abrogate proteoglycan synthesis by OA cartilage. Human OA cartilage was incubated with PGE2 (10 μM) with or without the COX-2-selective inhibitors celecoxib or rofecoxib (2 μM) for 6 days in serum-free medium. Medium was changed every 48 h. Proteoglycan synthesis was determined as 35S incorporation as described in Materials and Methods. The data represents mean ± SD (n = 5). ∗, p < 0.01 performed with five individual patient cartilage explants cultures.
PGE2 stimulates collagen degradation in OA cartilage in an MMP-dependent manner
Relative to normal cartilage, OA cartilage demonstrated elevated levels of collagen degradation as assayed by C1,2C fragment accumulation (1–4 ng/g cartilage) in the explant supernatants. C1,2C fragments were further increased in response to the addition of either PGE2 (40–60 ng/g cartilage) or IL-1β (30–40 ng/g cartilage; Fig. 3). To determine the role of MMP secretion in PGE2-augmented collagen degradation, we tested the effects of the broad-spectrum MMP inhibitor ilomastat on explants stimulated with PGE2. Addition of ilomastat (5 μg/ml) to PGE2-treated OA cartilage significantly inhibited collagen degradation (p < 0.02). Similar inhibition of collagen degradation was observed when cartilage explants were incubated with ilomastat (5 μg/ml) before stimulation with IL-1β. Taken together, these data suggest that PGE2-induced collagen degradation is mediated, at least in part, via synthesis/secretion of ilomastat inhibitable MMPs.

Type II collagen degradation in OA cartilage is augmented by PGE2. Human OA cartilage was incubated with or without PGE2 (10 μM), with or without the MMP inhibitor Ilomastat (5 μg/ml) for 72 h in serum-free medium. Degradation products of type II collagen were determined in the supernatant using the C1,2C ELISA kit. IL-1β served as positive control. The data represents mean ± SD (n = 3) performed with three individual patient cartilage explants cultures.
Type II collagen degradation in OA cartilage is augmented by PGE2. Human OA cartilage was incubated with or without PGE2 (10 μM), with or without the MMP inhibitor Ilomastat (5 μg/ml) for 72 h in serum-free medium. Degradation products of type II collagen were determined in the supernatant using the C1,2C ELISA kit. IL-1β served as positive control. The data represents mean ± SD (n = 3) performed with three individual patient cartilage explants cultures.
PGE2 regulates matrix gene expression in chondrocytes
We next analyzed the effect of PGE2 on expression of genes for MMPs, ADAMTS, and other extracellular matrix (ECM) proteins in chondrocytes. Chondrocytes were primed with PGE2 (10 μM) or treated with COX-2-selective inhibitor celecoxib (2 μM) 2 h before addition of IL-1β. Cells were collected and used for gene expression studies using TaqMan PCR. As shown in Fig. 4,A, PGE2 significantly inhibited the constitutive expression of the gene for aggrecan, but not type II collagen. PGE2 (10 μM) augmented IL-1β-induced MMP-13 and ADAMTS-5 expression (Fig. 4, B and C). In contrast, PGE2 inhibited IL-1β-induced MMP-1 expression and secretion (data not shown). To determine whether IL-1β-induced MMP-13 production was regulated by PGE2, we reproduced these experiments in the presence or absence of celecoxib (2 μM). As shown in Fig. 4 B, celecoxib inhibited IL-1β-induced MMP-13 gene expression and production in chondrocytes. These data suggest that PGE2, whether provided exogenously or secreted by chondrocytes in response to stimuli such as IL-1β, acts both to induce cartilage-degrading enzymes and to suppress matrix proteoglycan synthesis by chondrocytes.

A–C, PGE2 inhibits ECM protein synthesis and induces the matrix-degrading enzymes MMP-13 and ADAMTS5 in human OA chondrocytes. Chondrocytes were grown in monolayer culture for 48 h before stimulating with various pharmacological agents with or without PGE2 (10 μM) or IL-1β (10 ng/ml). Cells were harvested and relative baseline expression of type II collagen, aggrecan (A), MMP-13 (B), ADAMTS-5 (C), and GAPDH was determined by QPCR. Primary OA chondrocytes were then stimulated with PGE2 (10 μM), IL-1β (10 ng/ml), IL-1β + PGE2, or IL-1β + celecoxib (2 μM) for 24 h in serum-free medium. Supernatants and cells were harvested for MMP-13 (B) mRNA (QPCR) and protein expression using ELISA (R&D Systems).
A–C, PGE2 inhibits ECM protein synthesis and induces the matrix-degrading enzymes MMP-13 and ADAMTS5 in human OA chondrocytes. Chondrocytes were grown in monolayer culture for 48 h before stimulating with various pharmacological agents with or without PGE2 (10 μM) or IL-1β (10 ng/ml). Cells were harvested and relative baseline expression of type II collagen, aggrecan (A), MMP-13 (B), ADAMTS-5 (C), and GAPDH was determined by QPCR. Primary OA chondrocytes were then stimulated with PGE2 (10 μM), IL-1β (10 ng/ml), IL-1β + PGE2, or IL-1β + celecoxib (2 μM) for 24 h in serum-free medium. Supernatants and cells were harvested for MMP-13 (B) mRNA (QPCR) and protein expression using ELISA (R&D Systems).
EP4 receptors are overexpressed in OA cartilage
To determine which of the four EP receptors (EP1–EP4) were responsible for mediating PGE2 effects on cartilage, we first examined the expression of EP receptor genes in pooled samples of OA vs normal cartilage using Affymetrix microarray. Although all four EP receptor subtypes were expressed in both OA and normal cartilage, only EP4 was differentially overexpressed in cartilage from OA patients (data not shown). Differential overexpression of EP4 message was confirmed in three normal and five OA individual cartilage samples analyzed by TaqMan quantitative PCR (QPCR; Fig. 5).

Analysis of EP1, EP2, EP3, and EP4 expression in OA cartilage by TaqMan PCR. Differential up-regulation of EP4 receptor in OA cartilage was confirmed (TaqMan PCR) using three individual nonarthritic (autopsy) and 5 OA cartilage samples as described in Materials and Methods. The relative fold changes were calculated using 2-Δ CT method (21 ).
Analysis of EP1, EP2, EP3, and EP4 expression in OA cartilage by TaqMan PCR. Differential up-regulation of EP4 receptor in OA cartilage was confirmed (TaqMan PCR) using three individual nonarthritic (autopsy) and 5 OA cartilage samples as described in Materials and Methods. The relative fold changes were calculated using 2-Δ CT method (21 ).
Up-regulation of EP receptor expression was confirmed at the protein level using flow cytometric analysis of isolated OA chondrocytes. EP1–EP4 proteins were all present and identified on these cells. In contrast to our message studies in cartilage explants, expression of EP4 receptor protein was not found to be increased in isolated OA vs normal chondrocytes (data not shown); since chondrocyte isolation may result in loss of normal signals from ECM-derived proteins, subsequent experiments examining the role of EP4 and other EP receptors were performed using cartilage explants.
EP4 receptor regulate MMP secretion and ECM metabolism
As shown in Fig. 6,A, unstimulated OA cartilage explants spontaneously released proMMP-1 (8.7 ± 1.3 ng/g cartilage) and exogenous addition of PGE2 inhibited proMMP-1 (6.07 ± 1.36 ng/g cartilage, p < 0.03) production. Addition of an EP4 antagonist (AH23848) in the range of 10–50 μM but not EP2 antagonist (AH6809) augmented spontaneous proMMP-1 secretion (25.8 ± 7.8 ng/g) cartilage (p < 0.002; Fig. 6,A). Similarly, OA cartilage explants spontaneously released proMMP-13 (12.92 + 5.26 ng/g cartilage) and AH23848 (10–50 μM), but not AH6809, significantly inhibited spontaneous proMMP-13 secretion by >50–75% (p < 0.01; Fig. 6,B). Neither AH23848 nor AH6809 showed a dose-dependent effect on spontaneous proMMP-1 or proMMP-13 secretion in cartilage explant cultures. The maximum effect of inhibition or augmentation was observed at the 10 μM concentration. We further studied the effect of these antagonists on exogenous addition of PGE2-induced proMMP-1 and proMMP-13 secretion. AH23848 blocked the inhibitory effect of exogenous PGE2 on proMMP-1 (p < 0.01; Fig. 6,C) and augmentation of proMMP-13 (p < 0.01; Fig. 6,D). In contrast, an EP2 antagonist (AH6809) did not inhibit or augment PGE2-induced proMMP-13 and MMP-1 production (Fig. 6 D) respectively. These data suggest a unique role for EP4 in the regulation of chondrocyte metabolism in OA.

PGE2 augments MMP-13, but inhibits MMP-1 secretion via EP4 receptors. Human OA cartilage was incubated in the presence and absence of either AH23848 (10–50 μM) or AH23848 (10–50 μM) in 24-well plates in serum-free medium. Secretion of MMP-1 (A) and MMP-13 (B) were determined 24 h after stimulation in the culture supernatant. The data represents mean ± SD (n = 4) performed with four individual patient cartilage explants cultures. C and D, OA cartilage was incubated either with PGE2 (10 μM) with or without AH23848 (10 μM; EP4 antagonist) or AH6809 (EP2 antagonist) in serum-free medium. After 24 h, secretion of MMP-1 (C) and MMP-13 (D) were determined in the culture supernatants. The data represent mean ± SD (n = 3) performed with three individual patient cartilage explants cultures.
PGE2 augments MMP-13, but inhibits MMP-1 secretion via EP4 receptors. Human OA cartilage was incubated in the presence and absence of either AH23848 (10–50 μM) or AH23848 (10–50 μM) in 24-well plates in serum-free medium. Secretion of MMP-1 (A) and MMP-13 (B) were determined 24 h after stimulation in the culture supernatant. The data represents mean ± SD (n = 4) performed with four individual patient cartilage explants cultures. C and D, OA cartilage was incubated either with PGE2 (10 μM) with or without AH23848 (10 μM; EP4 antagonist) or AH6809 (EP2 antagonist) in serum-free medium. After 24 h, secretion of MMP-1 (C) and MMP-13 (D) were determined in the culture supernatants. The data represent mean ± SD (n = 3) performed with three individual patient cartilage explants cultures.
We next examined the role of PGE2 and EP4 receptors in mediating IL-1β effects on proteoglycan synthesis. Consistent with previous reports (19), IL-1β (1 ng/ml) inhibited proteoglycan synthesis (>50%; Fig. 7,A). Preincubation of OA cartilage with celecoxib or EP4 antagonist (AH23848) reversed IL-1β-mediated inhibition of proteoglycan synthesis by >70%. Whereas exogenous PGE2 reversed the protective effects of celecoxib on proteoglycan synthesis (Fig. 7 A), exogenous PGE2 had no effect on proteoglycan synthesis in the presence of EP4 antagonist (AH23848), confirming that PGE2-mediated proteoglycan suppression is also modulated via the EP4 receptor.
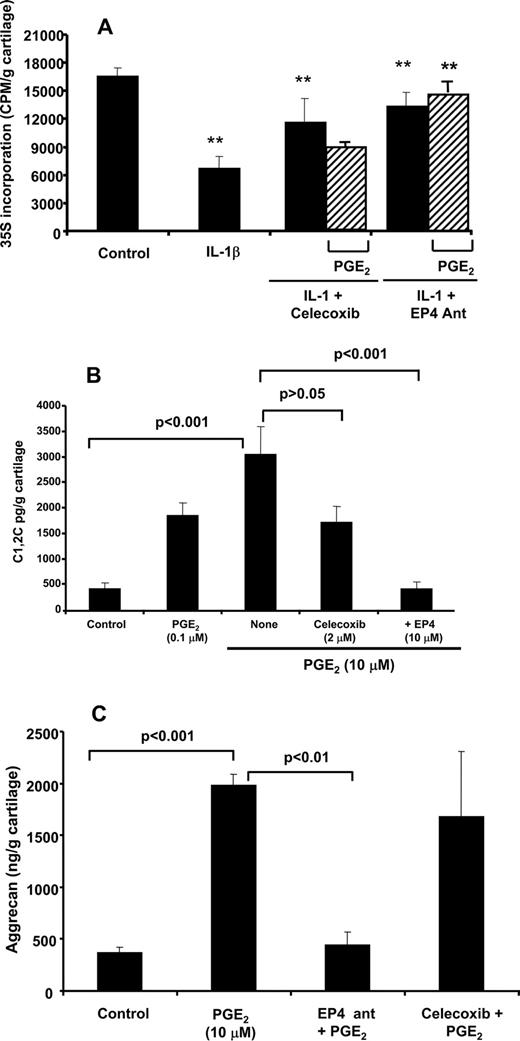
A, IL-1β-mediated inhibition of proteoglycan synthesis in OA cartilage is COX-2- and EP4 receptor-dependent. Human OA cartilage was incubated with PGE2 (10 μM) with or without celecoxib (2 μM), or AH23848 (10 μM) in the presence and absence of IL-1β in serum-free medium, before determination of proteoglycan synthesis as 35S incorporation. The data represent mean ± SD. ∗∗, p < 0.01 (n = 4) performed with four individual patient cartilage explants cultures. B and C, PGE2-mediated degradation of type II collagen degradation in human OA cartilage is EP4 dependent. Human OA cartilage was incubated with PGE2 (10 μM) with or without AH23848 (10 μM) in serum-free medium for 72 h (aggrecan; C) or 144 h for type II collagen degradation (B) were assayed as described in Materials and Methods. The data represent mean ± SD (n = 3) performed with three individual patient cartilage explants cultures.
A, IL-1β-mediated inhibition of proteoglycan synthesis in OA cartilage is COX-2- and EP4 receptor-dependent. Human OA cartilage was incubated with PGE2 (10 μM) with or without celecoxib (2 μM), or AH23848 (10 μM) in the presence and absence of IL-1β in serum-free medium, before determination of proteoglycan synthesis as 35S incorporation. The data represent mean ± SD. ∗∗, p < 0.01 (n = 4) performed with four individual patient cartilage explants cultures. B and C, PGE2-mediated degradation of type II collagen degradation in human OA cartilage is EP4 dependent. Human OA cartilage was incubated with PGE2 (10 μM) with or without AH23848 (10 μM) in serum-free medium for 72 h (aggrecan; C) or 144 h for type II collagen degradation (B) were assayed as described in Materials and Methods. The data represent mean ± SD (n = 3) performed with three individual patient cartilage explants cultures.
We also examined the role of EP4 in the regulation of collagen and aggrecan degradation. Addition of exogenous PGE2 (10 μM) resulted in increased levels of type II collagen fragments in OA cartilage (500 pg to 2–3 ng/g cartilage; Fig. 7 B). Preincubation of OA cartilage with EP4 antagonist (AH23848) (p < 0.001) but not celecoxib (p > 0.05) inhibited type II collagen degradation in response to addition of exogenous PGE2.
Preincubation of OA cartilage with EP4 antagonist (AH23848) also inhibited PGE2-mediated release of aggrecan fragments (452 ± 153 ng/g cartilage; Fig. 7 C). In contrast, celecoxib inhibited endogenous PGE2 production (>90% inhibition) and constitutive aggrecan release, but had no effect on aggrecan release in response to exogenously added PGE2 (1681 ± 629 ng/g cartilage). Thus, PGE2 regulates both collagen and aggrecan turnover via engagement of EP4 receptors.
Discussion
Relative to normal cartilage controls, OA cartilage expresses elevated levels of COX-2, with consequent increases in PGE2 production (11). In the current studies, we examined the consequences of PGE2 production in cartilage on matrix metabolism, demonstrating effects that are predominantly catabolic and mediated by signaling via the EP4 receptor. Although it has been appreciated for a decade that chondrocytes can up-regulate COX-2, relatively little is known about the consequences of COX-2-dependent PGE2 on OA cartilage metabolism. Several studies indicate that at the nano- to micromolar concentrations produced by OA tissues, PGE2 may exert catabolic effects (4, 24, 25, 26). For example, the selective COX-2 inhibitor celecoxib has been reported to reverse the decrease of matrix proteins, collagen type II, and aggrecan induced by mechanical stress (27), whereas the addition of PGE2 to cocultures of OA cartilage and synovial membrane abrogates the beneficial effects of COX-2 inhibitors on proteoglycan content (28). However, not all effects of PGE2 generation may be detrimental. Mastbergen et al. (29) reported that whereas COX-2-selective nonsteroidal anti-inflammatory drugs promote proteoglycan synthesis in cartilage explants, nonselective COX inhibitors promote proteoglycan degradation (29), suggesting the possibility of differential functions for COX-1- and COX-2-derived PGE2. The effects of PGs may also vary according to dose. In contrast to the nano/micromolar PGE2 doses used in our current studies, very low (picomolar) levels of PGE2 have been reported to inhibit proteoglycan degradation and enhance type II collagen and proteoglycan synthesis (30, 31). These results point to the subtlety of PGE2-driven chondrocyte metabolism in OA and the need for further characterization of PGE2-driven regulatory processes.
We further characterized the specific effects of nanomolar PGE2 on proteoglycan and type II collagen synthesis, confirming that PGE2 in this range contributes to both decreased matrix production and increased matrix degradation. We also demonstrated distinct and contrasting effects of PGE2 on expression/secretion of MMP-1 and MMP-13. Specifically, we observed that exogenous addition of PGE2 (1–10 μM) stimulated MMP-13 but inhibited MMP-1 expression and production, results similar to those previously reported in fibroblast-like synoviocytes (23). Increased MMP-13 expression was accompanied by increased cleavage of collagen and decreased proteoglycan synthesis in cartilage explant cultures. How the divergent synthesis of MMP-1 and -13 is regulated remains unknown, but we have previously reported that up-regulation of the nuclear orphan receptor NURR1 (NR4A2) in OA cartilage causes similar divergent effects, suggesting that the mechanism of PGE2 on MMP-1 and MMP-13 may be a result of NURR1 activation (NR4A2) (20).
One explanation for the ability of PGE2 to exert both anabolic and catabolic effects, depending upon dose and cell type, may relate to PGE2 receptor usage. Identification and characterization of the four known PGE2 receptors (EP1–4) has facilitated understanding of the role of each, providing opportunities to explore the pharmacological activities of PGE2, and to elucidate the specific effects of PGE2 in individual cell types. PGE2 receptors are coupled to distinct signaling pathways involving activation of phospholipase C (EP1), activation of adenyl cyclase (EP2 and EP4), and inhibition of adenyl cyclase (EP3) (17, 18), indicating their potential for divergent responses.
Of the four PGE2 receptors, only EP4 was up-regulated in OA vs normal cartilage. Our studies thus implicate EP4 in the mediation of PGE2 catabolic OA effects. In contrast, Sato et al. (12) reported that signaling via EP2 receptors suppresses MMP-13 induction in human OA chondrocytes. Thus, the disparate effects of PGE2 on chondrocyte function may reflect selective utilization of EP2 vs EP4 receptors. EP4 has been associated with promoting joint destruction in inflammatory arthritis. EP4 knockout mice exhibit reduced inflammation, incidence, and severity of collagen-induced arthritis. In contrast, homozygous deletion of the EP1, EP2, or EP3 receptors had no effect (30). In a recent study, PGE2 was shown to exacerbate collagen-induced arthritis in mice, acting via the inflammatory IL-23/IL-17 axis (31).
Our study is also the first to report that PGE2 augments IL-1β-induced ADAMTS-5 expression. ADAMTS-4 and -5 are both major aggrecanases involved in proteoglycan degradation, as well as potential targets for OA therapy (2, 32, 33). Although ADAMTS-4 and -5 are expressed constitutively in joint tissues and can efficiently digest aggrecan in vitro, the specific contribution of these enzymes to cartilage pathology is unknown. However, ADAMTS-5 null mice are protected against cartilage degradation in a model of inflammatory arthritis (34). Moreover, MMP-13 and ADAMTS-5 colocalize in OA cartilage (35) and increased expression of ADAMTS-5 has been observed in an ACL-transection dog model of early OA, as well as in focal areas of early degeneration in human cartilage (36, 37). Thus, the ability of PGE2 to up-regulate ADAMTS-5 strongly suggests a mechanism for PGE2-mediated proteoglycan degradation. The ability of IL-1β to induce both PGE2 production and ADAMTS–5 expressions suggest that PGE2 may act as a mediator of IL-1β-stimulated ADAMTS-5 up-regulation (38). Once again, RUNX2 may be involved, since both PGE2-induced expression of RUNX2 in mice bone marrow cells and overexpression of RUNX2 in chondrocytes led to up-regulation of MMP-13 and ADAMTS-5 (39, 40). Other mechanisms for PGE2-mediated cartilage degradation are not excluded by these observations. For example, Masuko et al. (41) have provided indirect evidence for the role of PGE2 in sphingosine-1-phosphate-mediated inhibition of aggrecan expression by human articular chondrocytes. The cAMP-increasing agents forskolin and IBMX have also been shown to exacerbate cartilage degradation in vivo (42). Since both EP2 and EP4 receptors can associate with and activate adenylyl cyclase (43), these provide potential mechanistic models for the effects of PGE2 on chondrocyte matrix homeostasis.
In summary, PGE2 produced by OA cartilage decreases proteoglycan synthesis and enhances the degradation of both aggrecan and type II collagen. These effects are associated with the up-regulation of MMP-13 and ADAMTS-5, two proteases which have been implicated in disease progression and which have been proposed as targets of disease modifying therapies (2, 32, 33). Our data further indicate that the catabolic effects of PGE2 are specifically mediated via engagement of the EP4 receptor. Together, these results suggest that PGE2 production is an underappreciated mediator of disease progression in OA and that selectively blocking its effects or targeting molecules such as COX-2, mPGES, EP4, and NURR1 could represent a strategy for OA disease modification.
Acknowledgments
We thank Jean Park for helpful discussion.
Disclosures
S.B.A. has served as a consultant to Pfizer Inc, Novartis, and Merck.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This work was supported in part by Grant R01-AR047206 from the National Institutes of Health Grant (to S.B.A.) and grants from the Joseph C. and Sophia Abeles Foundation, the Falk Family, and the Riley Family Foundation for their generous financial support for this research.
Abbreviations used in this paper: OA, osteoarthritis; MMP, matrix metalloproteinase; COX-2, cyclooxygenase 2; ECM, extracellular matrix; QPCR, quantitative PCR; mPGES, microsomal prostaglandin E synthase.
