

Abstract
The abundant serine proteinase inhibitor heparin cofactor II (HCII) has been proposed to inhibit extravascular thrombin. However, the exact physiological role of this plasma protein remains enigmatic. In this study, we demonstrate a previously unknown role for HCII in host defense. Proteolytic cleavage of the molecule induced a conformational change, thereby inducing endotoxin-binding and antimicrobial properties. Analyses employing representative peptide epitopes mapped these effects to helices A and D. Mice deficient in HCII showed increased susceptibility to invasive infection by Pseudomonas aeruginosa, along with a significantly increased cytokine response. Correspondingly, decreased levels of HCII were observed in wild-type animals challenged with bacteria or endotoxin. In humans, proteolytically cleaved HCII forms were detected during wounding and in association with bacteria. Thus, the protease-induced uncovering of cryptic epitopes in HCII, which transforms the molecule into a host defense factor, represents a previously unknown regulatory mechanism in HCII biology and innate immunity.
Introduction
Being the most abundant proteinase inhibitors in humans, serine proteinase inhibitors (serpins) regulate multiple proteolytic pathways in blood and tissues (1). Antithrombin III (ATIII) and heparin cofactor II (HCII), which are both present in human blood, inhibit proteases of the coagulation cascade (1, 2). Although current data indicate a pivotal role for ATIII in coagulation control, much less is known about the function of HCII. For example, although inherited deficiency of ATIII is clearly associated with thrombotic disorders, neither humans nor mice deficient in HCII present any evidence for thrombophilia under normal conditions (2–4). However, both ATIII and HCII interact with heparin and other glycosaminoglycans in a similar manner, undergoing significant conformational changes that facilitate protease inhibition by the common serpin mechanism (1, 5). In contrast to ATIII, however, HCII exploits its conformational mobility in a unique way. HCII has an unusual N-terminal acidic region, which, upon binding of HCII to glycosaminoglycans, is expelled from the molecules' cationic site, followed by proteolytic scission of the flexible N-terminal tail (6). Proteolysis of HCII by leukocyte elastase subsequently releases chemoattractant peptides derived from the N-terminal tail (7). Thus, although HCII shares similar overall conformational characteristics with other serpins, mounting evidence suggests that HCII may have evolved novel biological functions (1). For instance, although ATIII levels are reduced in almost all conditions associated with disseminated intravascular coagulation, reduced plasma levels of HCII are primarily detected during infection (8–11). Increasing evidence pointing toward a close link between hemostasis, proteolysis, and host defense (12–17) raised the hypothesis that HCII could play a role in host defense against infections.
Materials and Methods
Proteins, peptides, and patient materials
Human HCII was obtained from Enzyme Research Laboratories. The peptides KYE28 (KYEITTIHNLFRKLTHRLFRRNFGYTLR), GKS26 (GKSRIQRLNILNAKFAFNLYRVLKDQ), LKG23 (LKGETHEQVHSILHFKDFVNASS), SDP18 (SDPAFISKTNNHIMKLTK), and SGM22 (SGMKTLEAQLTPRVVERWQKSM) were synthesized by Biopeptide Co. (San Diego, CA) (>95% purity). Human acute and chronic wound fluids and chronic wound sloughs (CWS) were collected as previously described (18). The project was approved by the ethics committee, Lund University Hospital, and written consent was obtained from the patients.
Microorganisms and cells
Bacterial isolates of Escherichia coli ATCC 25922 and Pseudomonas aeruginosa 15159 were obtained from the Department of Bacteriology, Lund University Hospital, and cultured in Todd-Hewitt (TH) broth.
Degradation of HCII
HCII (13.5 μg, 0.6 μg/ml) was incubated with human leukocyte elastase (HLE) (0.3 μg, sp. act. 20 U/μg; Calbiochem) in a total volume of 45 μl 10 mM Tris (pH 7.4) at 37°C for 30 min. HCII degradation in wound fluid was assessed by incubation of wound fluid with different concentrations of HLE at 37°C for 1 h. The material was then subjected to SDS-PAGE under reducing conditions followed by immunoblotting.
Detection and analysis of HCII and HCII fragments
For sample separation with SDS-PAGE, 16.5% Tris-tricine gels (Bio-Rad) were used, followed by staining with Coomassie brilliant blue. For identification of HCII fragments in chronic leg ulcer wound fluids, 1.5 μl sample was analyzed by SDS-PAGE under reducing conditions. Proteins from mouse tissues were isolated using a protein purification kit (Total Protein Extraction Kit; Chemicon International). For detection of HCII during infection ex vivo, human or mouse blood or human citrate plasma was infected with P. aeruginosa 15159 (1:10 v/v inoculum/blood or plasma) grown overnight in TH broth. Samples were incubated on rotation, and for the blood samples, the material was centrifuged and plasma was prepared. Material incubated for 0, 2, 4, and 6 h was then subsequently subjected to analyses by SDS-PAGE and Western blot. Subsequent to separation by SDS-PAGE, proteins and peptides were transferred to nitrocellulose membranes (Hybond-C; GE Healthcare). Membranes were blocked by 3% (w/v) skimmed milk, washed, and incubated with polyclonal anti-human HCII (1:1000) or anti-mouse HCII Abs (1:1000; R&D Systems). After washing, HRP-conjugated secondary Abs (1:2000; DakoCytomation) were applied, and HCII bands were visualized by the Supersignal West Pico Chemiluminesent substrate developing system (Thermo Scientific). In pulldown assays, E. coli bacteria (1 to 2 × 109 CFU) were incubated with native or HLE-cleaved HCII for 2 h at 37°C. Heparin (10 mg/ml) was added for binding competition. After centrifugation, the bacterial pellet was washed with PBS, and bound proteins were eluted with 0.1 M glycine-HCl (pH 2). The pH of the eluted material was raised to 7.5 with 1 M Tris. Eluted proteins were precipitated by addition of 100 μl TCA to 400 μl sample, followed by incubation for 30 min on ice and centrifugation at 15,000 × g (4°C for 30 min). Precipitated material and supernatants were dissolved in SDS sample buffer and analyzed under reducing conditions followed by Western blot. For quantification of HCII in mouse plasma a commercial ELISA (USCN Life Science) was used.
LPS binding
Peptides (1, 2, and 5 μg) or proteins (2 and 5 μg) were bound to nitrocellulose membranes (Hybond-C; GE Healthcare). Membranes were blocked by 2% BSA in PBS (w/v) for 1 h at room temperature (RT) and subsequently incubated for 1 h with 125I-labeled LPS (0.13 × 106 cpm/μg) with or without unlabeled heparin (6 mg/ml). After incubation, membranes were washed with PBS, and the radioactivity was visualized using a Bas 2000 radioimaging system (Fuji).
Fast protein liquid chromatography
Fifteen micrograms native or digested HCII was subjected to fast protein liquid chromatography (ÄKTA purifier; GE Healthcare) using a HiTrap 1 ml Heparin HP column (Amersham Biosciences). After injection, samples were eluted with a linear gradient of 0–0.8 M NaCl in 10 mM Tris (pH 7.4).
HPLC
Peptide/protein fragments of HCII, digested with HLE, were separated by HPLC (Series 200; PerkinElmer) on a reversed-phase column (Brownlee Bio C18 5 μM 250 × 4.6 mm; PerkinElmer). After injection, samples were eluted with a gradient of acetonitrile in 0.1% aqueous trifluroacetic acid at 1 ml/min. Fractions were collected and stored at −80°C. Samples were freeze-dried, dissolved in water, and analyzed by radial diffusion assay SDS-PAGE, or for binding to 125I-labeled LPS by a slot blot assay.
Antimicrobial assays
Direct antimicrobial effects of HCII, HCIIa (HLE-cleaved HCII), or HCII-derived epitopes against E. coli ATCC 25922 and P. aeruginosa 15159 in 10 mM Tris HCl (pH 7.4) or by radial diffusion assay were determined as described earlier (13, 19). For determination of bacterial growth in citrated mouse blood, blood samples from HCII+/+or HCII knockout (HCII−/−) mice, or from HCII−/− mice supplemented with 100 μg/ml HCII, were infected with P. aeruginosa 15159 grown overnight in TH medium (1:10 v/v inoculum/blood, yielding an initial 1.7 × 106 CFU/ml at t = 0) and incubated under rotation at 37°C, and the number of CFU was determined after 6 h.
Flow cytometry analysis
E. coli bacteria (1 to 2 × 109 CFU) were incubated with buffer or native or cleaved HCII with or without heparin for 30 min at 37°C. The samples were then divided equally, centrifuged, washed, and resuspended in PBS with or without rabbit polyclonal Abs against human HCII. After incubation for 1 h at RT, bacteria were pelleted, washed, and incubated in PBS containing FITC-labeled goat anti-rabbit IgG Abs (1:500; Sigma-Aldrich) for 30 min at RT. The bacterial population was selected by gating with appropriate settings of forward scatter and side scatter. Flow cytometry analysis was performed using an FACSCalibur (BD Biosciences) and the Cell Quest acquisitions software (BD Biosciences).
Circular dichroism spectroscopy
Circular dichroism spectra were measured by a Jasco J-810 Spectropolarimeter (Jasco, Easton, MD). Measurements were performed in at least duplicate at 37°C in a 10-mm quartz cuvette under stirring with a peptide concentration of 10 μM. The effect on peptide secondary structure of LPS at a concentration of 0.2 mg/ml was monitored in the range of 200–260 nm. To account for instrumental differences between measurements, background subtraction was performed routinely. Signals from the bulk solution were also corrected for.
Electron microscopy
For transmission electron microscopy and visualization of HCII effects on bacteria, E. coli or P. aeruginosa (1 to 2 × 109 CFU/sample) was incubated for 2 h at 37°C in the absence or presence of heparin (10 mg/ml) and with: 1) HCII (8 μg) incubated with buffer or HLE (0.3 μg) for 30 min; or 2) human acute wound fluid (5 μl) incubated alone or supplemented with HLE (2.5 μg) for 1 h. To study HCII interactions with LPS, HCII was digested in 10 mM Tris (pH 7.4) as described in the first point above, followed by incubation with heparin or buffer (5 min at RT, 1:2 ratio) before the addition of LPS (1:1 ratio) for 5 min at RT. For the negative staining, suspensions were adsorbed onto 400 mesh carbon-coated copper grids and stained with 0.75% (w/v) uranyl formate as described previously (13). Fibrin slough from patients with chronic venous ulcers (CWS) was processed as recently described (13). The labeling of HCII was performed using polyclonal goat Abs against human HCII, in combination with gold-labeled EM rabbit anti-Goat IgG (H&L) and 20 nm Au (BB International). All samples were observed in an FEI Tecnai BioTWIN (North America NanoPort, Hillsbro, OR) transmission electron microscope operated at 60 kV accelerating voltage on a nitrogen-cooled stage (≈−140°C). Images were recorded with an Eagle 2k CCD camera (FEI) and the TIA acquisitions software. To quantify colocalization of HCII/HCIIa with LPS, a total of 300 HCII molecules in five different view fields were observed and presented as percentage binding.
Fluorescence microscopy
The impermeant probe FITC (6 μg/ml FITC; Sigma-Aldrich) was used to study membrane permeabilization of E. coli ATCC 25922 (1 × 107 CFU/ml) by HCII (3 μM), HLE-cleaved HCII (3 μM), HLE (0.2 μg/ml), or HCII-derived epitopes (30 μM) in 10 mM Tris (pH 7.4). The sample preparation and analysis was performed as described earlier (13).
Animal models
All animal experiments were approved by the Laboratory Animal Ethics Committee of Malmö/Lund, Sweden. The animals were purchased from the animal facility in Lund and housed under standard conditions of light and temperature with free access to laboratory chow and water. C57BL/6 wild-type (HCII+/+) and HCII−/− (3) mice were infected with P. aeruginosa 15159 bacteria either i.p. (2 × 108 CFU/mouse) or s.c. (0.4–1 × 109 CFU/mouse) or injected with 12 mg/kg E. coli LPS (O111:B4; Sigma-Aldrich). To study bacterial dissemination to target organs, spleen, liver, and kidney were harvested in PBS, placed on ice, homogenized, and subsequently CFUs were determined. Cytokines in blood, coagulation parameters, and HCII protein levels were determined 12–20 h after bacterial infection or LPS challenge.
Platelet analysis
Mouse blood (EDTA anticoagulated) was taken by cardiac puncture, and the number of platelets was determined with the VetScan HM5 System (Triolab).
Coagulation assays
All clotting times were analyzed using a coagulometer (Amelung, Lemgo, Germany). Activated partial thromboplastin time (aPTT) was measured by incubating 50 μl citrated plasma for 1 min followed by the addition of 50 μl Dapttin (Technoclone) for 200 s at 37°C. Clotting was initiated by the addition of 50 μl CaCl2 (30 mM). For the prothrombin time (PT), clotting was initiated by addition of 50 μl TriniClot-PT Excel (Trinity Biotech).
Cytokine assay
The levels of IL-6, IL-10, MCP-1, IFN-γ, and TNF-α were assessed using the Mouse Inflammation Kit (BD Biosciences) according to the manufacturer’s instructions.
Statistics
CFU data from several ex vivo or in vivo experiments were pooled, and the median is indicated. In other experiments, data are presented as mean from two to three experiments, and error bars indicate SEM. Statistical significance was evaluated with GraphPad Prism software 5.0 (GraphPad) using the Mann–Whitney U test with *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
Cleaved HCII binds to LPS and bacteria
We first set out to determine the properties of HCII and its cleaved form with focus on possible bacterial interactions. Digestion of pure HCII with human leukocyte elastase (HLE) yielded one major truncated HCII form with a m.w. ∼50 kDa, designated HCIIa below, which was promptly generated and appeared to be stable during the course of the digestion. Along with this, minor fragments of lower molecular weights were identified (Fig. 1A, Supplemental Fig. 1A–C). Notably, HCIIa exhibited significantly higher affinity for heparin in comparison with the intact molecule (Fig. 1B), which is consistent with previous reports (20, 21). Given the scission of the N-terminal, anionic fragment (having net charge [znet] −19) from HCII (total znet −5), calculations showed that the residual HCIIa was highly cationic (znet +14). Considering the observed increase in heparin affinity and charge, enhanced interactions with other anionic components, such as LPS, the main outer cell wall component of Gram-negative bacteria, seemed likely. In experiments addressing this, we found that in contrast to HCII, HCIIa indeed bound LPS, and the interaction was completely blocked by an excess of heparin (Fig. 1C). Correspondingly, FACS analyses demonstrated that only HCIIa bound to Gram-negative Escherichia coli bacteria, as illustrated by an increase in fluorescence intensity as compared with intact HCII, and that heparin inhibited this binding (Fig. 1D). Taking into account the presence of multiple minor fragments, including the possible release of the N-terminal tail of HCII (7), we set out to define the major HCII form bound to bacteria. By again exploiting the affinity to E. coli, this time using a pulldown assay (coprecipitation of HCII with bacteria), we identified the ∼50 kDa, HLE-releasable, HCIIa form as the major form bound, whereas intact HCII showed no affinity to bacterial cells (Fig. 1E). Compatible with the above results (Fig. 1C, 1D), heparin abolished the HCII binding to E. coli.

Proteolytically cleaved HCII binds to LPS and bacteria. (A) SDS-PAGE analysis of HCII subjected to HLE. Incubation times are indicated. (B) Comparison of heparin binding of HCII (black) and HLE-digested HCII (blue) using heparin affinity chromatography (fast protein liquid chromatography). (C) Slot blot analysis for detection of binding of HCII, HCIIa (HCII+HLE), and HLE to 125I-labeled LPS with or without heparin. (D and E) Analyses of interaction between HCII or HLE-cleaved HCII and E. coli bacteria in presence or absence of heparin. (D) The interaction between E. coli and intact and cleaved HCII was analyzed by flow cytometry. Top panel: bacteria incubated with intact HCII; bottom panel: bacteria incubated with cleaved HCII (HCII+HLE). Heparin blocked the binding of HCIa to the bacteria (bottom panel). Representative histograms are shown (n = 3; Control: E. coli only). (E) Evaluation of binding of intact and cleaved HCII to E. coli using pulldown assays. A representative Western blot is shown (n = 3). Heparin (Hep) abolished binding of HCIIa (HCII+HLE) to the pellet (P+Hep, rightmost lane). mAU, Milli-absorbance unit; P, bacterial pellet; S, supernatant.
Proteolytically cleaved HCII binds to LPS and bacteria. (A) SDS-PAGE analysis of HCII subjected to HLE. Incubation times are indicated. (B) Comparison of heparin binding of HCII (black) and HLE-digested HCII (blue) using heparin affinity chromatography (fast protein liquid chromatography). (C) Slot blot analysis for detection of binding of HCII, HCIIa (HCII+HLE), and HLE to 125I-labeled LPS with or without heparin. (D and E) Analyses of interaction between HCII or HLE-cleaved HCII and E. coli bacteria in presence or absence of heparin. (D) The interaction between E. coli and intact and cleaved HCII was analyzed by flow cytometry. Top panel: bacteria incubated with intact HCII; bottom panel: bacteria incubated with cleaved HCII (HCII+HLE). Heparin blocked the binding of HCIa to the bacteria (bottom panel). Representative histograms are shown (n = 3; Control: E. coli only). (E) Evaluation of binding of intact and cleaved HCII to E. coli using pulldown assays. A representative Western blot is shown (n = 3). Heparin (Hep) abolished binding of HCIIa (HCII+HLE) to the pellet (P+Hep, rightmost lane). mAU, Milli-absorbance unit; P, bacterial pellet; S, supernatant.
Cleaved HCII is antimicrobial
Whether binding to bacterial surfaces leads to direct bacterial killing was addressed by using E. coli bacteria for detection of possible antibacterial effects. As judged by radial diffusion assay, digestion of HCII with HLE yielded antimicrobial activity after 5 min digestion, and the effect was retained after extended incubation with the enzyme (Fig. 2A). Likewise, using viable counts, a direct antimicrobial activity was detected for cleaved HCII, but not for the intact form, at HCII concentrations close to physiological levels (≈1.5 μM) (11) (Fig. 2B). Similar results were obtained using Gram-negative P. aeruginosa bacteria (Supplemental Fig. 2A). Analyses after HPLC purification of cleaved HCII demonstrated that the isolated ∼50 kDa HCIIa molecule was of relatively high hydrophobicity, that it bound LPS, and was antimicrobial (Supplemental Fig. 1D), further linking the above antibacterial activities to HCIIa. At the ultrastructural level, only HCIIa caused disruption of bacteria, leading to disintegration and expulsion of cytoplasmic components (Fig. 2C). Bacteria incubated with HCIIa and heparin did not show any membrane damage and were similar to controls (Supplemental Fig. 2B). These results were similar to the bacterial destruction caused by membrane-acting antimicrobial peptides (12, 14, 15, 22). Correspondingly, studies using the impermeant fluorescent probe FITC demonstrated that only HCIIa caused membrane permeabilization of bacterial cells (Fig. 2D). Taken together, these results demonstrate that the generation of HCIIa by HLE is a prerequisite for binding to LPS and bacteria, bacterial permeabilization, and, ultimately, bacterial killing.

Proteolytically cleaved HCII exerts antimicrobial effects. (A) Evaluation of antimicrobial activity of HLE, HCII, or HCIIa (HCII+HLE) against E. coli bacteria using a radial diffusion assay. Inhibition zones (as illustrated above graph) were measured and presented as means ± SD (n = 3). (B) Antimicrobial effect of HCII preparations against E. coli in viable count assays. Bacterial survival (%) is shown as mean values ± SEM (n = 3). (C) Analysis of E. coli treated with HCII or HLE-cleaved HCII in 10 mM Tris (pH 7.4) using electron microscopy. (D) Fluorescence microscopy analysis of E. coli bacteria subjected to HCII, HCII+HLE, and HLE in 10 mM Tris (pH 7.4). Permeabilization was assessed using the impermeant probe FITC. Top panel: Nomarski images; bottom panel: fluorescence microscopy images of the same view fields (original magnification ×100).
Proteolytically cleaved HCII exerts antimicrobial effects. (A) Evaluation of antimicrobial activity of HLE, HCII, or HCIIa (HCII+HLE) against E. coli bacteria using a radial diffusion assay. Inhibition zones (as illustrated above graph) were measured and presented as means ± SD (n = 3). (B) Antimicrobial effect of HCII preparations against E. coli in viable count assays. Bacterial survival (%) is shown as mean values ± SEM (n = 3). (C) Analysis of E. coli treated with HCII or HLE-cleaved HCII in 10 mM Tris (pH 7.4) using electron microscopy. (D) Fluorescence microscopy analysis of E. coli bacteria subjected to HCII, HCII+HLE, and HLE in 10 mM Tris (pH 7.4). Permeabilization was assessed using the impermeant probe FITC. Top panel: Nomarski images; bottom panel: fluorescence microscopy images of the same view fields (original magnification ×100).
Proteolysis of HCII induces a conformational change facilitating LPS binding
The observations of a dramatic functional change of HCII upon cleavage by HLE, adding new LPS-binding and antibacterial capabilities to the molecule, and the fact that addition of heparin seemed to completely inhibit these effects suggested that release of the N-terminal acidic tail of HCII facilitates the uncovering of a heparin binding region of HCII, possibly by a conformational change, which subsequently mediates the host defense activities of HCIIa. Indeed, electron microscopy studies of single HCII molecules demonstrated that intact HCII presented a horseshoe-like structure, in contrast to the HLE-cleaved form, which assumed an almost stretched, linear shape (Fig. 3, top panel). Moreover, the majority of HCIIa forms (67 ± 12%) were found to bind LPS (thin fibrillar structures) (Fig. 3, middle panel, particularly clearly seen in the right panel insets). In contrast, only 18 ± 6% of the intact HCII molecules colocalized with LPS. Compatible with the above antibacterial experiments, the addition of heparin blocked the binding of LPS to HCIIa (11 ± 5% colocalization), similar to the levels observed for intact HCII and heparin (8 ± 4% colocalization) (Fig. 3, bottom panel), further indicating that a conformational change induced by proteolysis of HCII exposes a heparin- and LPS-binding region in the molecule.
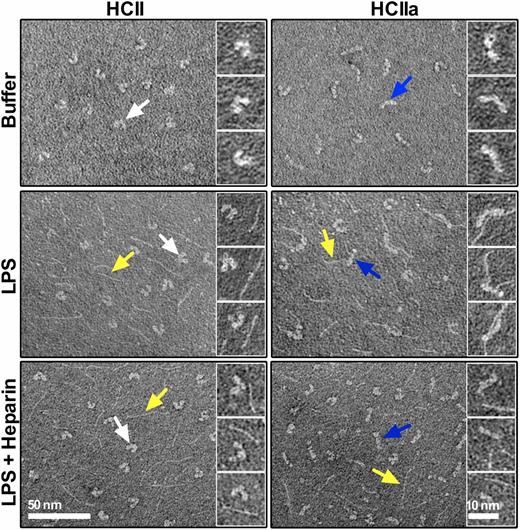
Cleavage of HCII induces a conformational change and LPS binding. Electron micrographs showing intact HCII or HLE-cleaved HCII (HCIIa), which were incubated with LPS or LPS with heparin or buffer only. Arrows: white, native HCII; blue, HCIIa; and yellow, LPS.
Cleavage of HCII induces a conformational change and LPS binding. Electron micrographs showing intact HCII or HLE-cleaved HCII (HCIIa), which were incubated with LPS or LPS with heparin or buffer only. Arrows: white, native HCII; blue, HCIIa; and yellow, LPS.
Definition of HCII epitopes mediating the antibacterial effects
HCII has two heparin-binding regions, located to the cationic and amphipathic helices A and D (6). To further define the epitopes responsible for the LPS-binding and antibacterial actions of intact HCIIa, peptides comprising those two helices, but also the other exposed helical regions of HCII (marked by different colors in Fig. 4A), were synthesized. Initial experiments using a slot-binding assay demonstrated that peptides from helix A and to a higher extent, helix D, bound LPS (Fig. 4B). The binding could be blocked by excess heparin (Fig. 4B, bottom panel), further verifying the overlap between heparin- and LPS-binding activities of HCII. These results corresponded to the LPS-binding data for HCIIa (Figs. 1C, 3). For both helix A and D peptides (GKS26 and KYE28, respectively), circular dichroism spectra revealed an induction of a helical conformation in the presence of E. coli LPS (Supplemental Fig. 3A). Furthermore, the helix A and D peptides were antibacterial in viable count assays using E. coli (Fig. 4C) and P. aeruginosa bacteria (Supplemental Fig. 3B), and it was notable that the doses required were compatible with those found to be effective for HCIIa-mediated killing of these bacteria. Finally, using E. coli, only peptides of helix A and D exerted permeabilizing effects on the bacteria (Fig. 4D). In summary, these data demonstrate that helix A and D mediate the LPS-binding and bactericidal effects of HCIIa.

Definition and functional analyses of HCII-derived peptides. (A) Illustration of HCII structure. Exposed helical peptide regions are indicated: helix A, GKS26 (green); helix D, KYE28 (orange); helix H, SGM22 (magenta); helix F, SDP18 (cyan); and helix C, LKG23 (yellow). (B) Slot blot assay for detection of binding of the indicated HCII-derived peptides to 125I-labeled LPS in the presence or absence of heparin. (C) Antimicrobial activity of HCII peptides against E. coli in 10 mM Tris, 0.15 M NaCl, with or without 20% citrate plasma. Bacterial survival (%) is presented as mean values ± SEM (n = 3). (D) Analysis of permeabilizing effects of HCII-derived peptides. E. coli bacteria were incubated with the indicated peptides and permeabilization assessed using the impermeant probe FITC. Top panel: Nomarski images; bottom panel: fluorescence microscopy images of the same view fields (original magnification ×100).
Definition and functional analyses of HCII-derived peptides. (A) Illustration of HCII structure. Exposed helical peptide regions are indicated: helix A, GKS26 (green); helix D, KYE28 (orange); helix H, SGM22 (magenta); helix F, SDP18 (cyan); and helix C, LKG23 (yellow). (B) Slot blot assay for detection of binding of the indicated HCII-derived peptides to 125I-labeled LPS in the presence or absence of heparin. (C) Antimicrobial activity of HCII peptides against E. coli in 10 mM Tris, 0.15 M NaCl, with or without 20% citrate plasma. Bacterial survival (%) is presented as mean values ± SEM (n = 3). (D) Analysis of permeabilizing effects of HCII-derived peptides. E. coli bacteria were incubated with the indicated peptides and permeabilization assessed using the impermeant probe FITC. Top panel: Nomarski images; bottom panel: fluorescence microscopy images of the same view fields (original magnification ×100).
HCII plays a role in protection against P. aeruginosa infection in vivo
To explore the physiological significance of the above findings, a series of experiments using HCII−/− and wild-type C57BL/6 (HCII+/+) mice were subsequently performed. First, we investigated whether a lack of HCII affects bacterial growth ex vivo. The results revealed that P. aeruginosa bacteria indeed showed enhanced growth in whole blood from HCII−/− mice when compared with blood from HCII+/+ mice. Addition of physiological amounts of HCII to blood from HCII−/− mice restored the bacterial numbers in blood to those observed for normal controls (Fig. 5A). Hypothetically, the effects of HCII, as described in Fig. 5A, could be related to the molecule’s inhibitory effect on thrombin and hence, interference with coagulation. However, coagulation assays showed a similar prolongation of the aPTT and PT in both groups, indicating a similar activation of the coagulation system in plasma derived from HCII+/+ and HCII−/− mice during infection ex vivo (Supplemental Fig. 4A). In separate experiments aimed at detecting HCII fragmentation during infection ex vivo, human or mouse blood was incubated with P. aeruginosa up to 6 h. Western blot analysis of samples taken every second hour showed that notwithstanding levels of intact HCII were reduced, no cleaved forms were detected in blood postinfection (Supplemental Fig. 4B). In contrast, HCII fragments were indeed detected, when human plasma was subsequently infected with P. aeruginosa (Supplemental Fig. 4C). Taken together, these results imply that bacteria-bound HCII is rapidly cleared in blood. Furthermore, the fact that HCII degradation occurred in (cell-free) plasma suggests that HCII is also sensitive to bacterial and/or other plasma-derived proteases.
![FIGURE 5. HCII suppresses bacterial growth ex vivo and in vivo. (A) P. aeruginosa was grown in blood from HCII+/+or HCII−/− mice or from HCII−/− mice supplemented with 100 μg/ml HCII [HCII−/−(+)]. The number of CFU at t = 0 was 1.7 × 106. After incubation for 6 h, the number of CFU was evaluated [HCII+/+, n = 19; HCII−/−, n = 18; HCII−/−(+), n = 13]. (B–E) HCII+/+ and HCII−/− mice were infected i.p. with P. aeruginosa, and analyses were performed after 12 h. Bacterial counts (HCII+/+, n = 17; HCII−/−, n = 21) (B), cytokines in plasma (HCII+/+, n = 8; HCII−/−, n = 8) (C), and platelet counts (HCII+/+, n = 17; HCII−/−, n = 18) (D) in comparison with noninfected mice (HCII+/+, n = 9; HCII−/−, n = 8). (E) Determination of aPTT and PT coagulation times (HCII+/+, n = 7; HCII−/−, n = 8). (F) Bacterial counts in organs of HCII+/+ and HCII−/− mice s.c. infected with P. aeruginosa (HCII+/+, n = 13; HCII−/−, n = 15). In bar diagrams, data are presented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. P. aer, P. aeruginosa.](https://aai.silverchair-cdn.com/aai/content_public/journal/jimmunol/190/12/10.4049_jimmunol.1203030/4/m_1203030fig05.jpeg?Expires=1716377398&Signature=KWGX1O8eWBItMdiIeK3-9nbfq4BWxS8CY~OKjGmHws-FWp4QiFzwjLM98WLhATchDAgXbKPxKoHcONFK6on-UdZAaVbeahSexeXWw3pYXkqrIAiqJQ7mGduwymF0Ch1RepH987giZ6HeuRAQpoUw781szSsPRrLqa2Sia6CZXfoawhbXGdyoUqxclughzvcpnaqq718HdOUuZ4t2vUcq0Csiz7Nw5~i76JF8hwDFluOOzkiQz276Lhs7m~K6aZP2W~7icSo55IctiuEp-dBv3UZ5Kt6pSk733Wi9nzksFY4bmeuGBabb6dfwFFIIml1lzrTh-XtrhSoa9Rbqt0NjDw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
HCII suppresses bacterial growth ex vivo and in vivo. (A) P. aeruginosa was grown in blood from HCII+/+or HCII−/− mice or from HCII−/− mice supplemented with 100 μg/ml HCII [HCII−/−(+)]. The number of CFU at t = 0 was 1.7 × 106. After incubation for 6 h, the number of CFU was evaluated [HCII+/+, n = 19; HCII−/−, n = 18; HCII−/−(+), n = 13]. (B–E) HCII+/+ and HCII−/− mice were infected i.p. with P. aeruginosa, and analyses were performed after 12 h. Bacterial counts (HCII+/+, n = 17; HCII−/−, n = 21) (B), cytokines in plasma (HCII+/+, n = 8; HCII−/−, n = 8) (C), and platelet counts (HCII+/+, n = 17; HCII−/−, n = 18) (D) in comparison with noninfected mice (HCII+/+, n = 9; HCII−/−, n = 8). (E) Determination of aPTT and PT coagulation times (HCII+/+, n = 7; HCII−/−, n = 8). (F) Bacterial counts in organs of HCII+/+ and HCII−/− mice s.c. infected with P. aeruginosa (HCII+/+, n = 13; HCII−/−, n = 15). In bar diagrams, data are presented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. P. aer, P. aeruginosa.
HCII suppresses bacterial growth ex vivo and in vivo. (A) P. aeruginosa was grown in blood from HCII+/+or HCII−/− mice or from HCII−/− mice supplemented with 100 μg/ml HCII [HCII−/−(+)]. The number of CFU at t = 0 was 1.7 × 106. After incubation for 6 h, the number of CFU was evaluated [HCII+/+, n = 19; HCII−/−, n = 18; HCII−/−(+), n = 13]. (B–E) HCII+/+ and HCII−/− mice were infected i.p. with P. aeruginosa, and analyses were performed after 12 h. Bacterial counts (HCII+/+, n = 17; HCII−/−, n = 21) (B), cytokines in plasma (HCII+/+, n = 8; HCII−/−, n = 8) (C), and platelet counts (HCII+/+, n = 17; HCII−/−, n = 18) (D) in comparison with noninfected mice (HCII+/+, n = 9; HCII−/−, n = 8). (E) Determination of aPTT and PT coagulation times (HCII+/+, n = 7; HCII−/−, n = 8). (F) Bacterial counts in organs of HCII+/+ and HCII−/− mice s.c. infected with P. aeruginosa (HCII+/+, n = 13; HCII−/−, n = 15). In bar diagrams, data are presented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. P. aer, P. aeruginosa.
To investigate a potential anti-infective role for HCII in vivo, HCII+/+ and HCII−/− mice were infected i.p. with P. aeruginosa bacteria, and bacterial dissemination was recorded in spleen, kidney, and liver (Fig. 5B). Analogously to the ex vivo experiments, significantly higher bacterial levels were observed in HCII−/− mice when compared with control mice. This was accompanied by significantly elevated levels of cytokines in mice deficient of HCII, particularly IL-6 and IFN-γ (Fig. 5C). A significant reduction in platelet counts (Fig. 5D), but no alterations in PT and aPTT, was observed in HCII−/− animals vis-à-vis controls (Fig. 5E). In an infection model using s.c. administration of bacteria, HCII−/− mice also displayed higher bacterial counts in the analyzed organs compared with wild-type animals (Fig. 5F). Thus, these results indicate that the major role of HCII in vivo is related to bacterial clearance and not coagulation control.
HCII levels are reduced and fragmented forms are present during infection in vivo
We further hypothesized that if HCII interacts directly with bacteria, the levels of HCII should be reduced during bacterial infection in vivo, analogously to the observations in infected blood ex vivo. Thus, plasma samples taken at different time points from C57BL/6 mice challenged with LPS, or infected with P. aeruginosa, were analyzed for HCII. Indeed, a reduction in plasma levels of HCII, as detected by Western blot (Fig. 6A) and ELISA (Fig. 6B), was observed over time. In the organs analyzed, reduced levels of HCII, particularly in kidney and liver, were also observed (Fig. 6C). We next investigated whether intact or proteolytically cleaved forms of HCII could be detected under inflammatory conditions, such as wounding. The results showed that intact HCII was found in wound fluids collected from patients with acute surgical wounds (Fig. 6D, left panel). Notably, addition of increasing amounts of HLE to these wound fluids, thus simulating conditions with excessive inflammation, such as those observed during infection, yielded fragments similar to the HLE-cleaved HCIIa forms described previously (Fig. 6D, middle panel) (21). Chronic wounds represent a clinical situation characterized by a high influx of neutrophils and excessive and uncontrolled levels of HLE (18, 23). Given this, wound fluids from these patients were investigated for HCII, and, as shown in Fig. 6D, right panel, these fluids contained mostly forms similar to HCIIa, indicative of a unique compartmentalization of HCIIa generation to specific microenvironments characterized by high inflammation. To further investigate the association of HCIIa with bacteria under physiological conditions, acute wound fluid was incubated with P. aeruginosa bacteria with or without HLE. Electron microscopy analysis using gold-labeled Abs directed against HCII demonstrated that elastase activation of HCII significantly increased its binding to bacteria (Fig. 6E, Supplemental Fig. 2C). Correspondingly, analysis of HCII in fibrin slough derived from a patient wound infected with P. aeruginosa showed that the molecule was indeed associated with bacterial surfaces (Fig. 6F), corresponding to the in vitro data presented in Fig. 1 on binding to bacteria. In summary, the data obtained from the mouse models indicate that the observed decrease, particularly of HCII plasma levels, is compatible with an extravascular consumption of HCII during infection and associated release of endotoxins, which is also compatible with observations of reduced HCII levels in humans during infection (8–10). Furthermore, the data with human wound fluids show that HCII, although present during wounding, is fragmented, particularly in response to excessive levels of HLE, conditions found in wounds characterized by high neutrophil influx and activation. This generated HCIIa associates with bacteria in human wound fluids ex vivo, as well as during wounding in vivo.

Detection of HCII during infection and inflammation. (A–C) C57BL/6 (HCII+/+) mice were injected i.p. with P. aeruginosa bacteria or E. coli LPS. Mice were sacrificed at indicated time points, and blood and organs were taken for analysis. (A) Western blot analysis of HCII in mouse plasma taken at the indicated time points from animals infected with P. aeruginosa or subjected to LPS. (B) Analysis of HCII levels in plasma of control mice and plasma of mice infected for 12 h with P. aeruginosa or challenged for 20 h with E. coli LPS. The data are presented relative to HCII levels in controls (n = 10, mean ± SEM is presented). (C) HCII levels in organs derived from mice either infected with P. aeruginosa (P. aer) or subjected to LPS. Tissues were collected at indicated times posttreatment, and detection was performed using Western blot analysis. β-actin levels in these tissues are shown for comparison (representative blots out of three experiments are shown). (D) Analysis of HCII in citrate plasma (CP) and various wound fluids by SDS-PAGE and Western blot using polyclonal Abs against HCII. Left panel: HCII detection in CP and six acute wound fluids (AWF, 1–6). Middle panel: AWF was incubated for 1 h with increasing amounts of HLE (indicated in μg) and analyzed for HCII. Right panel: Detection of HCII forms in six chronic wound fluid samples (CWF, 1–6). (E) AWF incubated with buffer or HLE was added to P. aeruginosa bacteria. HCII at bacterial surfaces was visualized using gold-labeled Abs (marked with ·). The mean number of gold particles (Au) per μm2 is indicated. (F) CWS: representative scanning electron micrograph of fibrin slough from chronic wounds stained for HCII with gold-labeled Abs (marked with ·). *p < 0.05, ***p < 0.001, Mann–Whitney U test. Con, plasma from noninfected mice.
Detection of HCII during infection and inflammation. (A–C) C57BL/6 (HCII+/+) mice were injected i.p. with P. aeruginosa bacteria or E. coli LPS. Mice were sacrificed at indicated time points, and blood and organs were taken for analysis. (A) Western blot analysis of HCII in mouse plasma taken at the indicated time points from animals infected with P. aeruginosa or subjected to LPS. (B) Analysis of HCII levels in plasma of control mice and plasma of mice infected for 12 h with P. aeruginosa or challenged for 20 h with E. coli LPS. The data are presented relative to HCII levels in controls (n = 10, mean ± SEM is presented). (C) HCII levels in organs derived from mice either infected with P. aeruginosa (P. aer) or subjected to LPS. Tissues were collected at indicated times posttreatment, and detection was performed using Western blot analysis. β-actin levels in these tissues are shown for comparison (representative blots out of three experiments are shown). (D) Analysis of HCII in citrate plasma (CP) and various wound fluids by SDS-PAGE and Western blot using polyclonal Abs against HCII. Left panel: HCII detection in CP and six acute wound fluids (AWF, 1–6). Middle panel: AWF was incubated for 1 h with increasing amounts of HLE (indicated in μg) and analyzed for HCII. Right panel: Detection of HCII forms in six chronic wound fluid samples (CWF, 1–6). (E) AWF incubated with buffer or HLE was added to P. aeruginosa bacteria. HCII at bacterial surfaces was visualized using gold-labeled Abs (marked with ·). The mean number of gold particles (Au) per μm2 is indicated. (F) CWS: representative scanning electron micrograph of fibrin slough from chronic wounds stained for HCII with gold-labeled Abs (marked with ·). *p < 0.05, ***p < 0.001, Mann–Whitney U test. Con, plasma from noninfected mice.
Discussion
Serpins are a superfamily of proteins using dramatic conformational changes, which enables them to control proteolytic events in various organisms. However, the serpin family includes also members that show no protease inhibitory activity. These serpins function in diverse biological processes, such as hormone transport (thyroxine-binding globulin) (24), blood pressure regulation (angiotensinogen) (25), tumor suppression (maspin) (26), and angiogenesis (pigment epithelium-derived factor) (27). In other cases, functional, inhibitory serpins display additional activities, as exemplified by C1 inhibitor (C1 INH), which shows anti-inflammatory effects including endotoxin binding (28), and protein C inhibitor (PCI), which exerts direct antimicrobial effects (29). In this perspective, the unique capacity of HCII to respond swiftly to proteases in inflammatory milieus, acquiring a conformation critical for interactions with bacteria and endotoxins, represents not only a conceptually novel function for this well-known serpin, but also constitutes a previously undisclosed host defense mechanism, employing a unique protease-dependent shape-shift.
From a structural perspective, several lines of evidence in this work indicate that the previously characterized helix A and D regions, known to interact with anionic glycosaminoglycans (6), mediate the antimicrobial effects of HCIIa. These two helices are both cationic and amphipathic (6), which are characteristics shared with many classical antimicrobial and LPS-interacting peptides such as LL-37 (22). Therefore, the current study implicates that the exposure of these two regions in the elongated HCIIa form underlies the binding of HCIIa to LPS and bacteria and its functional implications for bacterial clearance. In this context, it is notable that binding of C1 INH to LPS depends on N-linked glycosylation and on positively charged residues within the N-terminal nonserpin domain (28), a region that differs significantly from the highly anionic and chemotactic N terminus of HCII (6). With respect to PCI, the heparin-binding and antimicrobial epitope is assigned to the H helix of the molecule (29, 30). As demonstrated in Fig. 4, the corresponding region in HCII (peptide SGM22) showed neither LPS-binding nor antimicrobial activity. These observations, together with the fact that PCI shows remarkable protease stability (29) further imply a separate mechanism for PCI's action. Taken together, these observations indicate that during evolution, the innate immune functions of HCII, C1 INH, and PCI developed separately. Further, in contrast to the helical and antimicrobial LL-37 (31) and C-terminal thrombin peptides (12), found to be released from their holoproteins hCAP18 and thrombin, respectively, the herein described HCIIa form per se mediates the bactericidal and LPS-binding effects. In this perspective, the molecule therefore belongs to the class of larger antimicrobial proteins such as antimicrobial RNases (32), eosinophil cationic protein (33), lactoferrin (34), PCI (29), and histidine-rich glycoprotein (35), which all exert bactericidal effects, albeit without the need for the critical and unique enzymatic activation, followed by the dramatic shift in net charge, and the shape transition observed for HCII in this work.
From a functional point of view, the results do not exclude that the observed host defense role of HCII in vivo is mediated by additional mechanisms, active in parallel, either independently of bacteria, or as a result of the HCIIa–bacteria interaction. Indeed, the history of classical antimicrobial peptides and proteins illustrates that these molecules, initially attributed antimicrobial effects only, have since then been assigned multiple and diverse immunomodulatory roles (36). Hence, the in vivo effects of HCIIa observed in this study could go beyond direct antibacterial and LPS-binding effects and also encompass other more indirect effects dependent on the observed bacterial binding, such as enhanced phagocytosis, an assumption compatible with the observed reductions of HCII ex vivo and in vivo during infection. In summary, these results imply that HCIIa binding to bacteria, although being a prerequisite for its antibacterial action, is not the sole effector underlying the molecule's observed anti-infective role in vivo. Receptor-mediated clearance mechanisms have indeed been reported for serpin–enzyme complexes (37, 38), and inspired by these previous observations, present work addresses bacteria–HCIIa clearance in cell systems, ex vivo, and in vivo. These experiments however, are well beyond the scope of this present study focusing on the primary antibacterial role of HCII's unique shape-shift. Finally, considering the role of HCII as a thrombin inhibitor, reduced levels of HCII could lead to enhanced coagulation. However, analyses of the intrinsic as well as the extrinsic coagulation pathways during infection showed no such influence of HCII on these parameters, a finding compatible with the here proposed new role of HCII in innate immunity.
From a biological perspective, it may be envisioned that the protease-induced initial release of HCII's chemotactic N-terminal peptide (7), paired with shape- and charge-shifting of the residual main molecule, tunes HCII from an extracellular thrombin inhibitor to an activated host defense factor, thus facilitating a control of proteolytic as well as antibacterial activity in environments of localized and high inflammation. This capacity of HCII should be of particular importance during wounding and infection, situations in which an optimized host defense is critical for survival. The observed decreased levels of the molecule during infections in patients, paralleled by similar findings in animal models of endotoxic shock and severe P. aeruginosa infection also imply novel treatment possibilities for severe infections based on supplementation with HCII or its functional correlates.
Acknowledgements
We thank Ann-Charlotte Strömdahl and Maria Baumgarten for excellent technical assistance.
Footnotes
This work was supported by grants from the Swedish Research Council (projects 2009-3378 and 2009-3009); the Royal Physiographic Society in Lund; the Welander-Finsen, Knut and Alice Wallenberg, Crafoord, Österlund, and Kock Foundations; XImmune AB, The Swedish Government Funds for Clinical Research (Avtal om Läkarutbildning och Forskning), and a grant from the National Institutes of Health (R01-HL55520 to D.M.T.).
The online version of this article contains supplemental material.
Abbreviations used in this article:
- aPTT
activated partial thromboplastin time
- ATIII
antithrombin III
- C1 INH
C1 inhibitor
- CWS
chronic wound sloughs
- HCII
heparin cofactor II
- HCII−/−
heparin cofactor II knockout
- HLE
human leukocyte elastase
- PCI
protein C inhibitor
- PT
prothrombin time
- RT
room temperature
- serpin
serine proteinase inhibitor
- TH
Todd-Hewitt
- znet
net charge.
References
Disclosures
M. Malmsten and A.S. are founders of and own shares in XImmune AB, a company developing anti-inflammatory peptides for human therapy. The other authors have no financial conflicts of interest.
