

Abstract
Inflammatory diseases such as Aicardi–Goutières syndrome and severe systemic lupus erythematosus are generally lethal disorders that have been traced to defects in the exonuclease TREX1 (DNase III). Mice lacking TREX1 similarly die at an early age through comparable symptoms, including inflammatory myocarditis, through chronic activation of the stimulator of IFN genes (STING) pathway. In this study, we demonstrate that phagocytes rather than myocytes are predominantly responsible for causing inflammation, an outcome that could be alleviated following adoptive transfer of normal bone marrow into TREX1−/− mice. TREX1−/− macrophages did not exhibit significant augmented ability to produce proinflammatory cytokines compared with normal macrophages following exposure to STING-dependent activators, but rather appeared chronically stimulated by genomic DNA. These results shed molecular insight into inflammation and provide concepts for the design of new therapies.
Introduction
More than 20% of the United States adult population reportedly suffers from arthritis and other forms of autoimmune disease, including rheumatoid and juvenile arthritis, ulcerative colitis or Crohn’s disease, and systemic lupus erythematosus (SLE; characterized by high cytokine production) (1–3). The etiology of these diseases remains unknown. However, it is plausible that chronic infection by microorganisms or self-DNA released from necrotic or incompletely apoptosed cells, or possibly even cellular retrotransposons, may activate innate immune pathways that cause inflammation-aggravated autoimmunity (1–3). Recently, TREX1 mutations have been identified in patients with severe SLE and Aicardi–Goutières syndrome (AGS), which presents as a lethal neurologic syndrome with patients harboring high cytokine levels (4–8). TREX1 (DNase III) is a 3′→5′ DNA exonuclease that targets ssDNA as well as dsDNA (9). The ability of TREX1 to remove mismatched 3′ terminal deoxyribonucleotides at DNA strand breaks suggests that this exonuclease may serve in an editing role in DNA replication or gap filling during DNA repair (10–12). TREX1−/− mice do not exhibit any increase in spontaneous mutation rates, although surprisingly they suffer from inflammatory myocarditis of an autoimmune etiology resembling AGS, with the median lifespan for such animals being only 10 wk (13, 14). The endogenous substrate of TREX1 remains unclear. However, it was recently demonstrated that TREX1-deficient mice lacking the innate immune sensor stimulator of IFN genes (STING) completely alleviates TREX1-mediated disease and lethality in murine models (15). STING is an endoplasmic reticulum-associated, transmembrane-rich molecule that is activated by cytosolic DNA to trigger the production of numerous cytokines such as CXCL10 as well as type I IFN (16–18). STING may bind to DNA directly, and it can be activated through association with cyclic dinucleotides produced as second messengers from certain bacteria (19, 20). Additionally, a synthase referred to as cGAS (Mab-21 domain containing 1/C6orf150) is able to directly recognize cytosolic DNA and can produce cyclic dinucleotides (cyclic GMP–AMP [cGAMP]) similarly capable of facilitating STING activity and cytokine production (21–23). Inadvertent activation of STING by necrotic or inappropriately apoptosed cells is additionally known to cause self-DNA–triggered lethal inflammatory disease in mice (24).
Given these data, it is plausible to consider that TREX1 may serve as a negative regulator of STING to eliminate as yet unclarified STING ligands that would otherwise trigger chronic cytokine production and the observed lethal inflammatory disease. In this study, we demonstrate that a variety of TREX1-deficient hematopoietic cells exert high basal levels of STING-dependent cytokine production and are responsible for inflammation. Our results provide new insight into the molecular mechanisms underlying self-DNA–mediated disorders and highlight a new pathway that could be therapeutically targeted to alleviate such disease.
Materials and Methods
Mice
STING knockout mice (STING−/− [SKO]) were generated in our laboratory (16). TREX1+/− mice were provided by Dr. T. Lindahl. SKO mice were crossed with TREX1+/− mice to generate the double-knockout mice (STING−/−TREX1−/− [DKO]). Mice care and study were conducted under approval from the Institutional Animal Care and Use Committee of the University of Miami. Mouse genotypes from tail biopsies were determined using real-time PCR with specific probes designed for each gene by a commercial vendor (Transnetyx).
Primary cell culture
Mouse embryonic fibroblasts (MEFs) were obtained from embryos at embryonic day 15 by a standard procedure as described (16). Bone marrow–derived macrophages (BMDMs) and dendritic cells (BMDCs) were isolated from hindlimb femurs of 8- to 10-wk-old mice by a procedure as described (24). Recombinant mouse M-CSF (10 ng/ml; R&D Systems) and recombinant mouse GM-CSF (10 ng/ml; BioLegend) were used for the differentiation of CD11b+ and CD11c+ DCs.
Bone marrow transplantation
Bone marrow cells were primarily harvested from the long bones of donor mice by flushing with a 26-gauge needle. Single-cell suspensions were T cell depleted using anti-Thy1.2 (HO-13.4 supernatant) Ab at 1:5 (v/v) for 30 min on ice followed by addition of 12% Low-Tox-M rabbit complement (Cedarlane Laboratories) and incubation at 37°C for 1 h. Wild-type (WT) or TREX1−/− mice as recipients were irradiated with 650 rads from a 60Co source at a dose rate 44.5 cGy/min. On the next day, the mice were irradiated again at same condition. At 4 h after the second irradiation, recipient mice were injected i.v. with 5 × 106 T cell–depleted bone marrow cells obtained by flushing donor WT, TREX−/− (TKO), and STING−/−TREX1−/− (STKO) femurs. Four weeks after bone marrow transplants, ∼50–70% of chimerism was achieved.
Histopathology
Ten-week-old WT, TKO, STKO, and SKO mice were sacrificed and the heart tissues were fixed in 10% formalin for 48 h. All processes for paraffin block and H&E staining were performed at the pathology research resources histology laboratory in the University of Miami. Myocardial and pericardial inflammation scores were blind reviewed based on degree of focality and intensity (0, normal; 3, most severe). The sum of focality and intensity was used as a myocardial or pericardial inflammation score.
Abs
Rabbit polyclonal Ab against STING was developed in our laboratory as described previously (16). Other Abs used included TREX1 (611987; BD Biosciences), β-actin (A5441; Sigma-Aldrich), CD68 (MCA1957; Serotec), CD11c (bs-2508R; Bioss), ICAM1 (ab124759; Abcam), and CXCR3 (orb135585; Biorbyt).
Antinuclear Abs
For semiquantitative determination of antinuclear Abs (ANAs), the sera were incubated with HEp-2 tissue culture cells as a substrate (MBL International) according to the manufacturer’s instructions. The fluorescence was observed under a Leica SP5 spectral confocal inverted microscope.
Gene array analysis
Total RNA was prepared from the heart and BMDMs using an RNeasy Mini kit (74104; Qiagen, Valencia, CA). RNA quality was analyzed by a Bioanalyzer RNA 6000 Nano kit (Agilent Technologies, Santa Clara, CA). Gene array analysis was examined by an Illumina Sentrix BeadChip array (mouse WG6 version 2; Affymetrix, Santa Clara, CA) at the Oncogenomics Core Facility, University of Miami. Raw intensity values from Illumina array were uploaded on GeneSpring software from Agilent. Values were quantile normalized and log2 transformed to the median of all samples. Significantly differentially expressed genes were computed using the Student t test and selected using threshold of p ≤ 0.05. Hierarchical clustering and visualization of selected differentially expressed genes was performed on GeneSpring using the Pearson correlation distance method, and linkage was computed using the Ward method. Gene expression profiles were processed and statistical analysis was performed at the Sylvester Comprehensive Cancer Center Bioinformatics Core Facility at the University of Miami.
Quantitative real-time PCR
Total RNA was reverse transcribed using Moloney murine leukemia virus reverse transcriptase (Promega). Real-time PCR was performed using a TaqMan gene expression assay (Applied Biosystems) for innate immune genes and inflammatory cytokines (IFN-β, Mm010439546; TNF, Mm00443258; IL-6, Mm00446190; IL-1β, Mm01336189; CXCL10, Mm00445235; IFIT3, Mm0170846; ICAM1, Mm00516023; CXCR3, Mm1249867).
ELISA
BMDMs were seeded in 96-well plates at 3 × 104 cells/well and transfected using 4 μg/ml dsDNA90 and cGAMP and infected with HSV-γ34.5 (multiplicity of infection of 1) using Lipofectamine 2000 (11668019; Invitrogen, Carlsbad, CA) for 16 h. IFN-β levels were determined by a VeriKine mouse IFN-β ELISA kit (42400-1; PBL Assay Science, Piscataway, NJ). IL-1β, CXCL10, MCP3, IL-6, and TNF-α levels were determined using the a Procarta cytokine assay kit (Affymetrix) and analyzed on the Luminex 100 system.
Immunohistochemistry
Immunohistochemistry was performed on deparaffinized heart tissue. To expose target proteins, heat-induced Ag retrieval was performed using Borg Decloaker solution (Biocare Medical). Following Ag retrieval, tissues were blocked in normal goat serum (Vector S-1000) for 20 min at room temperature and incubated with primary Abs in Bond primary Ab diluents (AR9352). Tissues were washed with PBS and incubated for 30 min with ImmPRESS reagents (Vector Laboratories). Detection was performed using a DAB substrate kit for peroxidase (Vector Laboratories). Tissues were counterstained with hematoxylin and prepared for mounting.
BMDC synchronization
BMDCs were treated with 2 mM thymidine for 24 h. Thymidine was removed by washing with DMEM and adding fresh DMEM for 3 h to release cells. The cells were treated with 100 ng/ml nocodazole overnight, washed with DMEM, and harvested at 0, 8, and 24 h.
Fluorescence in situ hybridization for synchronized cells
Cells were fixed in 3.7% formaldehyde in 1× wash buffer (Dako, K5499) for 2 min at room temperature and washed twice. Fixed cells were dehydrated using 70, 85, and 96% ethanol for each step (2 min) and allowed to air dry for 15 min. The cells were co-denatured with an all mouse centromere probe (Kreatech, KI-30500G) at 75°C for 10 min and hybridized overnight at 37°C in a humidified chamber. On the next day, a stringent wash was performed following the procedure of a cytology fluorescence in situ hybridization (FISH) accessory kit (Dako) and mounted in ProLong antifade reagent (Molecular Probes). All samples were visualized with a Leica SP5 spectral confocal inverted microscope.
Statistical analysis
All statistical analyses were performed with a Student t test. Data were considered to be significantly different with p < 0.05.
Results
Cardiac-infiltrating leukocytes are the source of cytokine production in TREX1−/− mice
To further delineate the role of STING in TREX1-manifested disease, we examined the hearts from WT, SKO, TKO, or STKO mice. As previously demonstrated, TKO mice have enlarged, inflamed hearts and a median lifespan of ∼8–10 wk, an effect that was annulled by crossing such animals with mice that lack STING (Fig. 1A, 1B) (13). Microarray analysis of heart tissue indicated the presence of high levels of proinflammatory cytokines, such as TNF-α and IL-1β, in TKO mice compared with WT mice (Fig. 1C). Significantly, RNA representing these cytokines was dramatically reduced in the hearts of STKO mice, confirming that their production was indeed STING-dependent (Fig. 1C). Real-time PCR (RT-PCR) analysis confirmed the STING-dependent increase in proinflammatory cytokine levels in TKO mice (Fig. 1D). It was plausible that cardiomyocytes may be responsible for generating the observed proinflammatory cytokines. However, immunohistological analysis indicated that cardiomyocytes do not predominantly express STING (Fig. 1E). Because STING appears to control the pathway that upregulates these transcripts, we therefore surmised that cardiomyocytes are unlikely to be the major source of lethal cytokine production. Immunohistochemistry studies did indicate, however, that the hearts from TKO mice were heavily infiltrated with leukocyte-related cells such as macrophages, neutrophils, and DCs when compared with WT mice, as determined by immunohistochemistry using Abs to CD68 or CD11c (Fig. 1E). Although cardiomyocytes did not express STING, infiltrating immune cells were clearly validated as being able to express STING (Fig. 1E). This infiltration of leukocytes was absent in the hearts of STKO mice, suggesting that STING-generated cytokines were responsible for promoting the infiltration of such cells. Analysis of serum from the mice endorsed our RNA results and indicated the presence of high levels of circulating proinflammatory cytokines in TKO mice compared with WT mice that was similarly demonstrated to be dependent on STING (Fig. 1F). TKO mice were also observed to have high levels of ANAs compared with WT mice, the generation of which likewise appeared to be conditional on the presence of STING (Fig. 1G). To confirm the importance of infiltrating monocytes in the lethal production of proinflammatory cytokines, we perfused WT and TKO mice to reduce the presence of such cells in the tissue of the heart. Cytokine levels in perfused heart tissue were then reanalyzed by RT-PCR, as above. This study confirmed that cardiac tissue from perfused TKO mice had dramatically reduced proinflammatory cytokine levels (Fig. 1H). Thus, infiltrating macrophages and DC lineages and not cardiomyocytes are likely the major source of STING-dependent cytokine production.

Infiltrating immune-related cells cause STING-dependent proinflammatory gene induction in TKO mice. (A) Kaplan–Meier survival curve of TKO mice (n = 43) or STKO (n = 53) mice. Immunoblot of STING and TREX1 in heart from WT, TKO, SKO, and STKO mice. β-Tubulin was used as loading control. (B) Macroscopic view of heart of each genotype mice at the age of 10 wk. (C) Gene array analysis of hearts isolated from WT, TKO, STKO, and SKO mice at 10 wk age of mice. Total RNA was purified and transcripts analyzed by Illumina Sentrix BeadChip array (mouse WG6 version 2). The data discussed in this publication have been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus and are accessible through Gene Expression Omnibus series accession no. GSE59219 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE59219). (D) Quantitative RT-PCR (qPCR) analysis of TNF, IL-1β, IL-6, and IFN-β from heart as in (C). Data are shown as the means of four mice. (E) Immunohistochemical detection of STING, CD68, and CD11C in the hearts. Original magnification ×400. (F) Multiplex Luminex assay for IL-1β, TNF, CXCL10, and MCP-3 in the sera. Data are shown as the means of five mice. (G) ANAs in the sera from WT, TKO, STKO, and SKO mice. Original magnification ×1260. (H) qPCR analysis of IFN-β, IL-1β, and IL-6 in unperfused and perfused heart from 10-wk-old WT and TKO mice. Data are shown as the means of two mice. Error bars indicated SD. Statistical analysis was performed using a Student t test. *p ≤ 0.05, **p ≤ 0.005.
Infiltrating immune-related cells cause STING-dependent proinflammatory gene induction in TKO mice. (A) Kaplan–Meier survival curve of TKO mice (n = 43) or STKO (n = 53) mice. Immunoblot of STING and TREX1 in heart from WT, TKO, SKO, and STKO mice. β-Tubulin was used as loading control. (B) Macroscopic view of heart of each genotype mice at the age of 10 wk. (C) Gene array analysis of hearts isolated from WT, TKO, STKO, and SKO mice at 10 wk age of mice. Total RNA was purified and transcripts analyzed by Illumina Sentrix BeadChip array (mouse WG6 version 2). The data discussed in this publication have been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus and are accessible through Gene Expression Omnibus series accession no. GSE59219 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE59219). (D) Quantitative RT-PCR (qPCR) analysis of TNF, IL-1β, IL-6, and IFN-β from heart as in (C). Data are shown as the means of four mice. (E) Immunohistochemical detection of STING, CD68, and CD11C in the hearts. Original magnification ×400. (F) Multiplex Luminex assay for IL-1β, TNF, CXCL10, and MCP-3 in the sera. Data are shown as the means of five mice. (G) ANAs in the sera from WT, TKO, STKO, and SKO mice. Original magnification ×1260. (H) qPCR analysis of IFN-β, IL-1β, and IL-6 in unperfused and perfused heart from 10-wk-old WT and TKO mice. Data are shown as the means of two mice. Error bars indicated SD. Statistical analysis was performed using a Student t test. *p ≤ 0.05, **p ≤ 0.005.
TREX1−/− monocytes chronically produce high background levels of proinflammatory cytokines
Data indicate that TREX1 prevents the activation of STING by as yet unclarified mechanisms (15). Thus, it is conceivable that cells lacking TREX1 may possibly be more sensitive to cytosolic DNA–dependent innate immune signaling because STING activity could conceivably be enhanced. To evaluate this, we transfected TREX1-deficient MEFs with dsDNA or cGAMP or infected such cells with the DNA pathogen HSV-1. These experiments indicated that TREX1-deficient MEFs did not generate appreciably more type I IFN when stimulated with STING ligands or after infection with HSV-1 (Supplemental Fig. 1A–C). A similar effect was noticed when examining BMDMs or BMDCs that lacked Trex1 (Fig. 2A). In fact, TREX1-deficient macrophages and DCs appeared to generate less type I IFN following stimulation at least with many STING ligands. Subsequently, we also confirmed that TREX1-deficient macrophages did not generate significantly higher levels of cytokines compared with their WT counterparts following engulfment of apoptotic or necrotic cells (Supplemental Fig. 1D). Thus, loss of TREX1 does not necessarily render cells more sensitive to the production of cytokines, in comparison with normal macrophages or DCs, after STING-dependent stimulation. Interestingly, we did notice that unstimulated macrophages or DCs lacking TREX1 constitutively generated more STING-inducible genes compared with WT cells. To evaluate this further, we isolated peritoneal macrophages and PBMCs from TKO mice and confirmed by RT-PCR that such cells did appear to generate higher levels of innate immune proteins such as IFIT3 and CXCL10, compared with WT cells, in a STING-dependent manner under resting conditions (Fig. 2B, 2C). DNA microarray analysis confirmed that BMDMs from TREX1-deficient mice exhibited augmented proinflammatory gene induction under nonstimulated conditions that was not observed in the absence of STING (Fig. 2D). These observations were again confirmed by RT-PCR analysis (Fig. 2E–G). Thus, TREX1-deficient monocytes generate higher background levels of STING-dependent proinflammatory cytokines compared with normal cells.

TREX1-deficient macrophages and DCs generate high background levels of proinflammatory cytokines. (A) ELISA of IFN-β in BMDMs and BMDCs treated with 4 μg/ml dsDNA90 (ISD) and cGAMP and infected with HSV-γ34.5 (multiplicity of infection of 1). Results are from one representative of at least two independent experiments. Lipo, lipofectamin only; UI, uninfected. (B) qPCR of IFIT3 and CXCL10 in macrophages isolated from WT, TKO, STKO, and SKO mice peritoneal cavities. (C) qPCR of IFIT3 and CXCL10 in PBMCs. (D) Gene array analysis by Illumina Sentrix BeadChip array (mouse WG6 version 2) from total RNA purified in WT, TKO, and STKO BMDMs treated with or without dsDNA90. The data discussed in this publication have been deposited in National Center for Biotechnology Information’s Gene Expression Omnibus and are accessible through Gene Expression Omnibus series accession no. GSE59219 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE59219). (E) Immunoblots show genotypes in BMDMs, BMDCs, and PBMCs using anti-STING and anti-TREX1. qPCR is shown of IFIT3 and CXCL10 mRNA level in BMDMs (F) and BMDC (G). All data are the means of duplicates for at least two mice. Error bars indicated SD. Statistical analysis was performed using a Student t test. **p ≤ 0.005.
TREX1-deficient macrophages and DCs generate high background levels of proinflammatory cytokines. (A) ELISA of IFN-β in BMDMs and BMDCs treated with 4 μg/ml dsDNA90 (ISD) and cGAMP and infected with HSV-γ34.5 (multiplicity of infection of 1). Results are from one representative of at least two independent experiments. Lipo, lipofectamin only; UI, uninfected. (B) qPCR of IFIT3 and CXCL10 in macrophages isolated from WT, TKO, STKO, and SKO mice peritoneal cavities. (C) qPCR of IFIT3 and CXCL10 in PBMCs. (D) Gene array analysis by Illumina Sentrix BeadChip array (mouse WG6 version 2) from total RNA purified in WT, TKO, and STKO BMDMs treated with or without dsDNA90. The data discussed in this publication have been deposited in National Center for Biotechnology Information’s Gene Expression Omnibus and are accessible through Gene Expression Omnibus series accession no. GSE59219 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE59219). (E) Immunoblots show genotypes in BMDMs, BMDCs, and PBMCs using anti-STING and anti-TREX1. qPCR is shown of IFIT3 and CXCL10 mRNA level in BMDMs (F) and BMDC (G). All data are the means of duplicates for at least two mice. Error bars indicated SD. Statistical analysis was performed using a Student t test. **p ≤ 0.005.
Adoptively transferred normal bone marrow rescues TREX1−/− mice
To further evaluate the role of hematopoietic cells in TREX1-manifested disease, we retrieved bone marrow cells (BMCs) from 6-wk-old WT or TKO mice and adoptively transplanted them into 6-wk-old irradiated TKO mice. We observed that <50% of TKO mice receiving TKO BMCs survived past 10 wk, whereas, significantly, little to no mortality was observed in TKO mice receiving WT BMCs (Fig. 3A). However, 6-wk-old WT mice receiving TKO BMCs but not WT BMCs exhibited noticeable mortality (∼25%) during the same period (Fig. 3B). The body weight of WT mice receiving TKO BMCs also decreased compared with WT mice receiving WT BMCs (Fig. 3C). The introduction of WT BMCs into TKO mice similarly rescued the loss of body weight typically found in mice lacking Trex1 (Fig. 3D). Thus, adoptively transferred BMCs from WT mice can increase the life expectancy of animals with defective TREX1 function, emphasizing the important role that hematopoietic cells alone play in the manifestation of AGS-like disease.

Elimination of TREX1-deficient hematopoietic cells alleviates inflammatory cytokine production in TKO mice. Survival curves (A and B) and body weight (C and D) of TKO or WT mice transplanted with WT, TKO, or STKO bone marrow. STKO BM > TKO, TKO mice reconstituted with STKO bone marrow (n = 4);TKO BM > TKO, TKO mice reconstituted with TKO bone marrow (n = 4); TKO BM > WT, WT mice reconstituted with TKO bone marrow (n = 7); WT BM > TKO, TKO mice reconstituted with WT bone marrow (n = 4); WT BM > WT, WT mice reconstituted with WT bone marrow (n = 5). Results are shown in Kaplan–Meier curve with a mean of at least four mice from three independent experiments. Error bars indicated SEM. Statistical analysis was performed using a Student t test. x-Axes indicate weeks after bone marrow transplant. *p ≤ 0.05. (E) Representative H&E staining (HE) of heart tissue (left, original magnification ×400) and ANA assay with the sera (right, original magnification ×1260). (F) Inflammation score (left) and semiquantitation of fluorescent intensity (right) from (E). (G) Multiplex Luminex assay for IL-6, TNF, and MCP-3 in the sera from WT BM > WT (n = 4) and TKO BM > WT (n = 2) mice. (H) qPCR analysis of IFIT3 and mRNA in PBMCs from the mice same as (A). Error bars indicated SD. Statistical analysis was performed using a Student t test. *p ≤ 0.05, **p ≤ 0.005.
Elimination of TREX1-deficient hematopoietic cells alleviates inflammatory cytokine production in TKO mice. Survival curves (A and B) and body weight (C and D) of TKO or WT mice transplanted with WT, TKO, or STKO bone marrow. STKO BM > TKO, TKO mice reconstituted with STKO bone marrow (n = 4);TKO BM > TKO, TKO mice reconstituted with TKO bone marrow (n = 4); TKO BM > WT, WT mice reconstituted with TKO bone marrow (n = 7); WT BM > TKO, TKO mice reconstituted with WT bone marrow (n = 4); WT BM > WT, WT mice reconstituted with WT bone marrow (n = 5). Results are shown in Kaplan–Meier curve with a mean of at least four mice from three independent experiments. Error bars indicated SEM. Statistical analysis was performed using a Student t test. x-Axes indicate weeks after bone marrow transplant. *p ≤ 0.05. (E) Representative H&E staining (HE) of heart tissue (left, original magnification ×400) and ANA assay with the sera (right, original magnification ×1260). (F) Inflammation score (left) and semiquantitation of fluorescent intensity (right) from (E). (G) Multiplex Luminex assay for IL-6, TNF, and MCP-3 in the sera from WT BM > WT (n = 4) and TKO BM > WT (n = 2) mice. (H) qPCR analysis of IFIT3 and mRNA in PBMCs from the mice same as (A). Error bars indicated SD. Statistical analysis was performed using a Student t test. *p ≤ 0.05, **p ≤ 0.005.
After 12 wk, hearts from the transplanted mice were retrieved and analyzed. A slight increase in the sizes of hearts retrieved from irradiated WT mice was noted following their transplantation at 6 wk with TREX1-deficient BMCs (TKO) but not WT BMCs (data not shown). H&E analysis further confirmed significant infiltration of leukocytes within the hearts of WT mice transplanted with TKO BMCs, but not in irradiated WT mice receiving WT BMCs (Fig. 3E). A greater inflammation score was similarly observed in the WT mice receiving TKO BMCs (Fig. 3F). Significantly, irradiated TKO mice receiving WT BMCs had fewer infiltrating leukocytes and exhibited less cardiac-related inflammation when compared with TKO mice receiving TKO BMCs (Fig. 3E, 3F). Depletion studies confirmed the copious infiltration of CD11b-expressing cells in the hearts of TKO mice (Supplemental Fig. 2A). It is thus plausible that monocytes such as macrophages and DCs are alone intrinsically capable of manifesting TREX1-associated inflammatory disease.
We next analyzed the serum of the transplanted mice. This study indicated that irradiated WT mice receiving TKO BMCs, but not WT BMCs, also exhibited high levels of ANAs similar to Trex1-deficient mice (Fig. 3E, 3F). Conversely, TKO mice receiving WT BMCs had markedly reduced levels of ANAs, as determined by fluorescence microscopy (Fig. 3E, 3F). Normal mice receiving TKO BMCs but not WT BMCs also acquired high levels of proinflammatory cytokines such as IL-6 and TNF-α in their serum (Fig. 3G). Increased levels of STING-activated genes including IFIT3 were further noted in PBMCs from WT mice receiving TKO BMCs (11 wk after transplantation), confirming that transplanted BMCs maintained their ability generate high levels of innate immune–related proteins, likely responsible for transferring inflammatory symptoms to normal mice (Fig. 3H). Collectively, we conclude that monocytes intrinsically produce abnormally high basal levels of proinflammatory cytokines in a STING-dependent manner in the absence of Trex1.
Intrinsic nuclear DNA aggravates STING-dependent signaling in TREX1−/− monocytes
The mechanisms underlying the above-detailed events remain unclear. It is possible that TREX1-deficient mice may be more susceptible to virus infection, but no significant infections were observed, and TREX1-deficient cells and mice have actually been shown to be more resistant to virus infection compared with their WT counterparts (25, 26). It is also possible that in the absence of TREX1, cells of the hematopoietic lineage may be more sensitive to resident microbes. However, antibiotic treatment of TREX1−/− mice treated from 4 wk of age mice did not reduce evidence of myocarditis or eliminate inflammatory symptoms during an 8-wk period of treatment (Supplemental Fig. 2B). Also note that macrophages from TKO animals did not appear to be more sensitive to cytokine production compared with their WT counterparts following stimulation with viruses (Fig. 2A, Supplemental Fig. 1A).
It has been proposed that TREX1-deficient mice may be susceptible to retrotransposon activation (14). Plausibly, TREX1 may play a housekeeping role in eliminating endogenous retroviruses that may be activated and that may stimulate innate immune signaling pathways (14, 27). To evaluate this, we developed a non-PCR–based assay based on NanoString technology that had probes able to detect 10 full-length retroviruses that are present in the mouse genome (28–30). However, we were not able to detect significant retrovirus activity in either macrophages or DCs isolated from TREX1-deficient mice (Supplemental Fig. 3). Although this does not rule out that retroviruses may not play a role in TREX1-manifested disease, there does not appear to be significant retroviral presence at least in cells of these types as determined using this technology. Thus, microbes were unlikely to be responsible for the observed aggravation of TREX1-deficient monocytes.
Neutrophils have been reported to exert antimicrobial properties by extruding their DNA into extracellular netlike traps. The cells then die in a process referred to as NETosis (31, 32). Conceivably, the exposure of DNA to the cytosol in cells undergoing NETosis may activate STING, especially in the absence of TREX1. However, we did not see enhanced NETosis in neutrophils lacking TREX1 compared with WT cells, nor did TREX1-deficient cells undergoing NETosis appear to produce more proinflammatory cytokines in mice (Supplemental Fig. 4). Thus, NETosis would not appear to be a major cause of Trex1-mediated disease.
It has additionally been reported that TREX1 may play a role in facilitating the cell division processes. For example, Trex1-deficient cells show a defective G1/S transition and chronic ATM-dependent checkpoint activation (10). Significant amounts of ssDNA molecules produced during S the phase were noted to accumulate in the endoplasmic reticulum, where STING and Trex1 normally reside. Thus, it is plausible that Trex1 may perform some housekeeping role important for degrading aberrant DNA species that may be generated through the genome replication process, which is capable of escaping the nucleus with the consequence of potentially activating cytosolic DNA innate immune sensors such as STING. To address this possibility, we retrieved conventional DCs from WT or TKO mice and synchronized them with thymidine and nocodazole that arrests cells in the G2 phase. Approximately 20% of cells appeared to be arrested in G2 following this treatment (Fig. 4A). After removal of the drug, cells were monitored for progression through the cell cycle, and the mRNA of key cytokines was measured. This analysis indicated that WT BMDCs treated in this way exhibited little cytokine production throughout cell division. However, BMDCs retrieved from TKO mice exhibited greater cytokine production as they progressed through the cell cycle, which eventually tapered off (Fig. 4B). FISH studies using labeled genomic DNA indicated the strong presence of cytosolic DNA species in the cytoplasm of BMDMs lacking TREX1 as well as in WT BMDMs (Fig. 4C). However, only in TREX1-deficient cells was enhanced cytokine production noted. Collectively, our data demonstrate that TREX1 may be responsible for eliminating erroneous DNA produced through the cell division process that may escape into the cytoplasm.
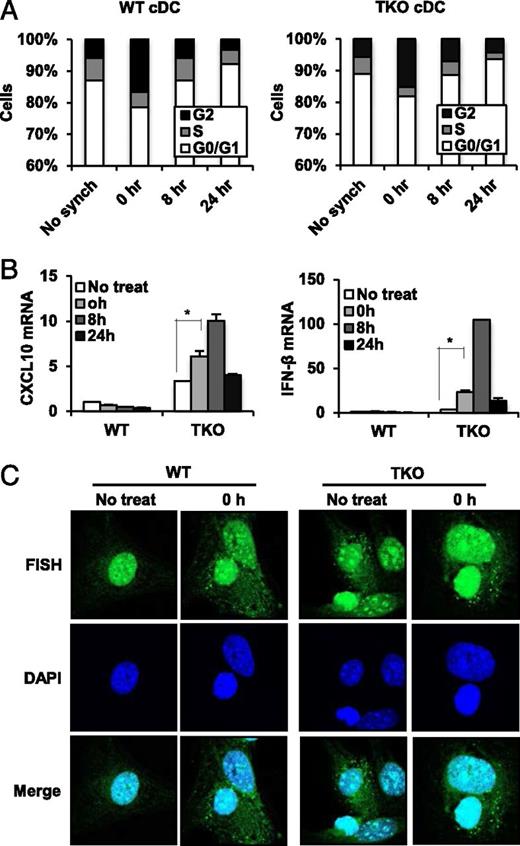
DNA species left over from cellular replication activates STING in Trex1 deficiency. (A) Cell cycle analysis in BMDCs isolated from WT and TKO mice at 0, 8, and 24 h after release from thymidine/nocodazole block for synchronization. (B) qPCR analysis of IFIT3 and IFN-β in BMDCs isolated from WT and TKO mice at 0, 8, and 24 h after release from thymidine/nocodazole block for synchronization. Error bars indicated SD. Statistical analysis was performed using a Student t test. *p ≤ 0.05. (C) FISH of all mouse centromere in BMDMs isolated from WT and TKO mice at 0 h after release from synchronization. DAPI was used for counterstaining. The cells were examined under Leica SP5 spectral confocal inverted microscope. Original magnification ×1260. Data are representative of at least two independent experiments.
DNA species left over from cellular replication activates STING in Trex1 deficiency. (A) Cell cycle analysis in BMDCs isolated from WT and TKO mice at 0, 8, and 24 h after release from thymidine/nocodazole block for synchronization. (B) qPCR analysis of IFIT3 and IFN-β in BMDCs isolated from WT and TKO mice at 0, 8, and 24 h after release from thymidine/nocodazole block for synchronization. Error bars indicated SD. Statistical analysis was performed using a Student t test. *p ≤ 0.05. (C) FISH of all mouse centromere in BMDMs isolated from WT and TKO mice at 0 h after release from synchronization. DAPI was used for counterstaining. The cells were examined under Leica SP5 spectral confocal inverted microscope. Original magnification ×1260. Data are representative of at least two independent experiments.
The upregulation of adhesion molecules attracts monocytes to cardiac tissue
Whereas TREX1-deficient APCs appear to be basally active, or “smoldering,” plausibly due to aberrant DNA species generated during the cell cycle that are able to activate STING, it is unclear why such cells predominantly infiltrate the cardiac region. However, it is possible that cytokines produced from TREX1-deficient APCs may in turn activate myocyte- and/or endothelial-generated chemokines and adhesion molecules that would, in turn, attract TKO monocytes producing appropriate receptors. Principally, we noted that monocytes lacking TREX1 produced high levels of the chemokines CCL4, CCL5, CXCL9, and CXCL10, among others, in a STING-dependent manner (Figs. 2D, 5A). Subsequently, we noted that the receptors of many of these chemokines (such as ICAM1, VCAM1, and CXCR3), the production of which can be stimulated by TNF-α and IL-1, were also upregulated in the heart tissue of TKO mice, as determined by RT-PCR (Fig. 5A–C). ICAM1 and VCAM1 bind to integrins such as CD11a and CD11b, found on selected hematopoietic cells, including macrophages and neutrophils. Such integrins play a key role in leukocyte adhesion and migration (Fig. 5D). Because infiltrating TKO APCs attracted to these regions express high levels of CCL4, CCL5, CXCL9, and CXCL10, which are, in turn, powerful chemoattractants for monocytes/macrophages, T cells, NK cells, and DCs, it would be anticipated that they would positively and potently promote further APC infiltration. Thus, basally activated monocytes from TKO mice are a major cause of inflammatory disease.

ICAM-1 and CXCR3 in endothelial cells of heart attract inflammatory cells in TKO mice. (A) Fold changes of chemokines in TKO and STKO compared with WT macrophages from gene array analysis in Fig. 2D and fold changes of chemokine receptors and adhesion molecules in TKO and STKO heart in comparison with WT heart from gene array analysis in Fig. 1C. qPCR of ICAM-1 (B) and CXCR3 (C) in unperfused and perfused heart (p-WT and p-TKO) from 10-wk-old WT and TKO mice same as Fig. 1H. Error bars indicated SD. Statistical analysis was performed using a Student t test. *p ≤ 0.05, **p ≤ 0.005. (D) Immunohistochemical detection of ICAM1 and CXCR3 in the hearts of WT and TKO mice. Cell percentages were blind reviewed by a pathologist. Original magnification ×400.
ICAM-1 and CXCR3 in endothelial cells of heart attract inflammatory cells in TKO mice. (A) Fold changes of chemokines in TKO and STKO compared with WT macrophages from gene array analysis in Fig. 2D and fold changes of chemokine receptors and adhesion molecules in TKO and STKO heart in comparison with WT heart from gene array analysis in Fig. 1C. qPCR of ICAM-1 (B) and CXCR3 (C) in unperfused and perfused heart (p-WT and p-TKO) from 10-wk-old WT and TKO mice same as Fig. 1H. Error bars indicated SD. Statistical analysis was performed using a Student t test. *p ≤ 0.05, **p ≤ 0.005. (D) Immunohistochemical detection of ICAM1 and CXCR3 in the hearts of WT and TKO mice. Cell percentages were blind reviewed by a pathologist. Original magnification ×400.
Discussion
In conclusion, we demonstrate in the present study that TREX1-mediated lethality in mice is largely due to inflammatory myocarditis caused by activated, infiltrating macrophages, neutrophils, and DCs. These TREX1−/− smoldering APCs exhibited pronounced expression of maturation/activation markers such as CD68 (macrophages) and CD11c (DCs) compared with WT cells. TREX1−/− macrophages and DCs generated elevated levels of proinflammatory cytokines in vitro and were predominantly responsible for the production of cytokines such as IL-1β, TNF-α, and IL-6 in the serum of TREX1−/− mice. Such cytokines are known to trigger the production of leukocyte binding adhesion molecules and chemoattractants, including members of the integrin family (ICAM, VCAM) and CXCR3 in cardiac myocytes, fibroblasts, and endothelial cells (33, 34). The production of these adhesion molecules and chemoattractants would, in turn, serve to attract smoldering TREX1−/− macrophages and DCs (35). However, other organs are known to exhibit signs of inflammatory damage, presumably due to similar reasons, namely, the possible upregulation of leukocyte attracting molecules (14). Significant expression of CXCL9 and CXCL10 was observed to be produced by TREX1−/− monocytes, which can further attract and activate a variety of leukocytes as well as induce the activation of selected T cell subsets (36). In this light it has been shown that T cells can contribute toward TREX1-mediated disease (15). Thus, APCs that lack TREX1 appear to express high background levels of proinflammatory cytokines and chemokines that can induce lethal cardiomyositis (34, 36).
The cause of cytokine production in TREX1−/− hematopoietic cells remains to be fully clarified. However, treatment of mice with antibiotics did not alleviate TREX1-mediated lethality, indicating that microbiota/normal flora were not largely responsible for inducing APC activation. Indeed, we observed that TREX1-deficient macrophages did not express elevated levels of proinflammatory cytokines in response to cytosolic DNA, bacteria, necrotic cells, or apoptotic cells compared with normal cells. Rather, monocytes lacking TREX1 appeared to exhibit high background levels of cytokine production in vitro, in the absence of any noticeable stimuli. These data suggest that such APCs are activated in an “intrinsic” manner, perhaps by self-DNA or even endogenous retroviruses. However, we did not observe any significant retroviral activity in macrophages or DCs lacking TREX1, as determined using a non-PCR–based NanoString assay (10, 14, 28). TREX1-deficient cells have been reported to suffer defects in the cell cycle progression, a consequence that led to the reported accumulation of ssDNA in the endoplasmic reticulum of such cells (10). Using FISH analysis, we did observe that TREX1-deficient DCs and macrophages exhibited high levels of leaked DNA into the cytoplasm during cell cycle progression. It is possible that TREX1 is responsible for degrading inappropriately copied or damaged DNA that might be aberrantly produced during genomic replication (10, 11). Such DNA that leaks into the cytoplasm may be expected to activate the DNA-sensing pathway controlled by STING, leading to the elevated production of proinflammatory cytokines. It is noteworthy that STING is predominantly present in cells of the hematopoetic lineage and endothelial cells but not in myocytes or several other types of tissue. Thus, such cells, generally considered to be terminally differentiated, would be expected to escape producing cytokines through leaked DNA in a STING-dependent manner. As mentioned, it is known that other tissues, aside from the heart, also exhibit inflammation in mice defective in TREX1, possibly through upregulating adhesion molecules (15). Human patients suffering from AGS are not known to develop inflammatory myocarditis because presumably they die of other causes prior to noticeable myocardial inflammatory events becoming evident (2). It thus remains to be seen whether smoldering monocytes occur in human patients with AGS or severe SLE that could be responsible for lethal encephalitis.
Finally, we show that adoptive transfer of normal bone marrow into irradiated TREX1-deficient mice, even after 6 wk of age, could reduce inflammation and extend survival. Conversely TREX1−/− bone marrow-derived cells could induce inflammation in irradiated WT mice. Aside from indicating that exogenously nonstimulated TREX1-deficient cells are predominantly responsible for proinflammatory disease, our data thus suggest that adoptive transfer therapy may be useful for patients suffering from TREX1-manifested AGS and SLE. Finally, our data provide molecular insight into the causes of self-DNA–mediated inflammatory disorders and afford new targets that could plausibly be therapeutically controlled to help prevent such diseases.
Acknowledgements
We thank Delia Gutman and Auristela Rivera for maintaining the mice, Dr. Tomas Lindahl and Dr. Deborah E. Barnes for TREX1+/− mice, Dr. Tianli Xia for mERV NanoString CodeSet design, Dr. Biju Issac of the Sylvester Comprehensive Cancer Center Bioinformatics Core Facility for gene expression array analysis, Dr. Robert Levy and Robert Newman for helping with bone marrow transplants, and Dayami Hernandez for helping with immunohistochemistry.
Footnotes
The dataset presented in this article has been submitted to the National Center for Biotechnology Information's Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE59219.
The online version of this article contains supplemental material.
Abbreviations used in this article:
- AGS
Aicardi–Goutières syndrome
- ANA
antinuclear Ab
- BMC
bone marrow cell
- BMDC
bone marrow–derived dendritic cell
- BMDM
bone marrow–derived macrophage
- cGAMP
cyclic GMP–AMP
- DC
dendritic cell
- DKO
double-knockout
- FISH
fluorescence in situ hybridization
- MEF
mouse embryonic fibroblast
- qPCR
quantitative real-time PCR
- RT-PCR
real-time PCR
- SKO
STING−/− (knockout)
- SLE
systemic lupus erythematosus
- STING
stimulator of IFN genes
- STKO
STING−/−TREX1−/− (knockout)
- TKO
TREX−/− (knockout)
- WT
wild-type.
References
Disclosures
The authors have no financial conflicts of interest.
