

Abstract
Aberrant Wnt/β-catenin signaling occurs in several inflammatory diseases, including inflammatory bowel disease and inflammatory bowel disease–associated colon carcinogenesis. However, its role in shaping mucosal immune responses to commensals in the gut remains unknown. In this study, we investigated the importance of canonical Wnt signaling in CD11c+ APCs in controlling intestinal inflammation. Using a mouse model of ulcerative colitis, we demonstrated that canonical Wnt signaling in intestinal CD11c+ APCs controls intestinal inflammation by imparting an anti-inflammatory phenotype. Genetic deletion of Wnt coreceptors, low-density lipoprotein receptor–related proteins 5 and 6 (LRP5/6) in CD11c+ APCs in LRP5/6ΔCD11c mice, resulted in enhanced intestinal inflammation with increased histopathological severity of colonic tissue. This was due to microbiota-dependent increased production of proinflammatory cytokines and decreased expression of immune-regulatory factors such as IL-10, retinoic acid, and IDO. Mechanistically, loss of LRP5/6-mediated signaling in CD11c+ APCs resulted in altered microflora and T cell homeostasis. Furthermore, our study demonstrates that conditional activation of β-catenin in CD11c+ APCs in LRP5/6ΔCD11c mice resulted in reduced intestinal inflammation with decreased histopathological severity of colonic tissue. These results reveal a mechanism by which intestinal APCs control intestinal inflammation and immune homeostasis via the canonical Wnt-signaling pathway.
Introduction
The Wnt-signaling pathway is critical for development of the intestine and for maintaining gut homeostasis (1, 2). The Wnt-signaling cascade has been known to play an important role in the development and differentiation of immune cells (3). The low-density lipoprotein receptor–related proteins 5 and 6 (LRP5/6) are key mediators of the canonical Wnt-signaling pathway (1, 2). Aberrant Wnt signaling occurs in several inflammatory diseases, including inflammatory bowel disease (IBD) and IBD-associated colon cancer (1, 2, 4). Although the focus of most research has been directed toward how the Wnt-signaling cascade regulates intestinal stem cell proliferation and epithelial cell maintenance, as well as its effects on cancer initiation and progression, its role in shaping mucosal immune responses to commensal flora in the gut remains largely unknown. In addition, the molecular mechanism by which the Wnt-signaling pathway in APCs regulates intestinal inflammation is still undefined.
Gut microbial communities have profound effects on human health, and loss of immune tolerance to gut microflora is inextricably linked to IBD and several other autoimmune diseases (5, 6). The gut microbiota plays an important role in shaping mucosal innate and adaptive immune responses (7, 8). Intestinal APCs such as dendritic cells (DCs) and macrophages (MPs) play a pivotal role in mediating mucosal tolerance and suppressing inflammation to commensal microflora (9–11). Previous studies in tumor settings have shown that the canonical Wnt-signaling pathway programs DCs into a regulatory state and promotes immune tolerance to tumors (12–15). In inflammatory disease settings, in contrast, recent studies have shown that the Wnt-signaling pathway acts as a counter-regulatory mechanism to control chronic inflammation and restore homeostasis (16–19). Wnt ligands in the gut environment can also act on immune cells and shape mucosal immune responses to commensal flora. Therefore, we hypothesized that canonical Wnt signaling in intestinal APCs is critical for maintaining a balance between immune tolerance and immune response to commensal flora in the gut. We also hypothesize that this is critical for suppressing intestinal inflammation and maintaining gut homeostasis.
In this study, we demonstrate that canonical Wnt signaling in CD11c+ APCs suppresses colonic inflammation in murine models of ulcerative colitis (UC). Canonical Wnt signaling imparts a regulatory phenotype on colonic APCs, which in turn induces regulatory T cell (Treg) differentiation while limiting the differentiation of pathological effector T cells. Conditional deletion of the coreceptors LRP5 and LRP6 in CD11c+ APCs in LRP5/6ΔCD11c mice resulted in an altered microbial compartment in the colon and enhanced susceptibility to intestinal inflammation. This is due to a microbiota-dependent increase in the expression of inflammatory cytokines and altered T cell homeostasis in the colon. Furthermore, our study demonstrates that, in LRP5/6ΔCD11c mice, conditional activation of β-catenin in CD11c+ APCs resulted in reduced intestinal inflammation and a reduction in histopathological severity of the colonic tissue.
Materials and Methods
Mice
C57BL/6 and CD11c-cre mice (20) were originally obtained from Jackson Laboratory and bred on-site. LRP5-floxed (LRP5FL) mice and LRP6-floxed (LRP6FL) mice were originally provided by Dr. B.O. Williams (Van Andel Research Institute, Grand Rapids, MI) and were cross-bred to generate homozygous LRP5/6-floxed (LRP5/6FL) mice as previously described (13, 18, 21). LRP5/6FL mice were crossed to transgenic mice expressing Cre recombinase under the control of the CD11c promoter (Jackson Laboratory) to generate mice in which LRP5/6 (LRP5/6ΔCD11c) were deficient in CD11c+ APCs (13, 18). Successful Cre-mediated deletion was confirmed by PCR and protein expression analyses, as in our previous studies (13, 18). LRP5/6FL and LRP5/6ΔCD11c mice were caged together. β-Catflox(ex3) mice were originally provided by Makoto Taketo (Kyoto University Graduate School of Medicine, Japan) and were cross-bred to CD11c-cre mice to generate mice expressing active β-catenin specifically in CD11c+ APCs (Act-βcatCD11c mice) (18, 22). Act-βcatCD11c mice were then bred to LRP5/6ΔCD11c mice to generate LRP5/6ΔCD11c X Act-βcatCD11c mice. All experiments were carried out with age-matched controls unless specified otherwise. All mice were housed under specific pathogen-free conditions at Augusta University, with animal care protocols approved by the Institutional Animal Care and Use Committee.
Abs and reagents
Abs against mouse CD3 (145-2C11), CD4 (GK1.5), CD8a (53-6.7), CD45 (30-F11), Foxp3 (FJK-16s), IL-10 (JES5-16E3), CD11c (N418), CD11b (M1/70), I-Ab (25-9-17), CD64 (X54-5/7.1), F4/80 (BM8), CD90.1 (HIS51), V α 2 TCR (B20.1), V β 5.1/5.2 TCR (MR9-4), IFN-γ (XMG1.2), and IL17A (17B7) were purchased from eBioscience. LRP5, LRP6, nonphosphorylated active β-catenin, β-catenin, and β-actin Abs were obtained from Cell Signaling Technology. CD11c and CD11b microbeads were purchased from Miltenyi Biotec.
Induction of colonic inflammation
Colonic inflammation was induced as previously described (23). Briefly, mice were subjected to one cycle of dextran sodium sulfate (DSS) treatment, whereby mice were given DSS (36–50 kDa) in their drinking water (at a dose as indicated in the 18Results) for 7 d followed by 8 d of normal drinking water. Some mice were subjected to antibiotic treatment prior to initiation of DSS administration. Mice were monitored for weight change, diarrhea, and rectal bleeding as previously described (23, 24). Diarrhea was scored as: 0, normal stool; 1, soft but formed pellet; 2, very soft pellet; 3, diarrhea (no pellet); or 4, dysenteric diarrhea. Rectal bleeding was recorded as: 0, no bleeding; 2, presence of occult blood in stool; or 4, gross macroscopic bleeding.
Leukocyte preparation and flow cytometry
Lamina propria (LP) leukocytes from colons were isolated as described in our previous study (25). Isolated LP leukocytes were collected, washed, and stained with Abs specific for mouse CD4 and Foxp3 and analyzed by FACS. Briefly, single-cell suspensions from lymph nodes, spleen, and LP were resuspended in PBS containing 5% FBS. After incubation for 15 min at 4°C with the blocking Ab 2.4G2 (anti-FcγRIII/I), the cells were stained with the appropriately labeled Abs. Samples were then washed twice in PBS containing 5% FBS. In some experiments, mononuclear cells from colonic LP or spleen were cultured with PMA (50 ng/ml) plus ionomycin (750 ng/ml) in the presence of GolgiStop and GolgiPlug for 6 h. The cells were then stained for CD4 followed by intracellular staining of IFN-γ, IL-17A, and IL-10. In other experiments, cell pellets obtained from EDTA washes of the colons were collected and used to quantify relative Wnt gene expression in epithelial cells.
Antibiotic treatment of mice
Antibiotic treatment of mice was performed as described in our previous studies (24, 25). In brief, LRP5/6FL and LRP5/6ΔCD11c mice were fed with an antibiotic mixture (1 g/l ampicillin, 1 g/l metronidazole, 1 g/l neomycin sulfate and 0.5 g/l vancomycin) in drinking water for 6 wk. All antibiotics were purchased from Sigma-Aldrich.
Ex vivo colon culture and ELISAs
Biological triplicates of 1 cm–long sections of the ascending colon were excised, removed of feces, washed three times with sterile HBSS, and then longitudinally opened, as previously described (23, 24). Colon sections were then placed into culture in complete RPMI 1640 media (supplemented with l-glutamine, penicillin, streptomycin, tetracycline, and 2% FBS) and cultured for 2 d at 37°C with 5% CO2. Supernatants were then collected, and cytokine concentrations were determined by ELISA. IL-17, IL-6, IL-10, IL-22, TNF-α, IFN-γ, and IL-1β were quantitated using BD Biosciences ELISA kits.
Colonic APC sorting
CD11c+ APCs were positively selected from the leukocyte preparation by elution via MACS LS columns (Miltenyi Biotec) and then FACS sorted for DCs (CD45+I-Ab+CD11c+ F4/80−CD64−) and MPs (CD45+I-Ab+CD11b+F4/80+CD64+) (26). After sorting, DCs or MPs (105) were cultured in 0.2 ml RPMI 1640 complete medium in 96-well round-bottom plates. Cell culture supernatants were analyzed after 48 h for indicated cytokine production by ELISA.
OT-II CD4+ T cell adoptive transfer
Naive CD4+CD25− T cells were isolated from the spleen and lymph nodes of Rag1−/− OT-II Thy1.1 transgenic mice and 5 × 106 cells were intravenously transferred into wild-type floxed (WT-FL) and LRP5/6ΔCD11c mice. Mice received 5 mg OVA by gavage on 5 consecutive d after transfer.
Bacterial DNA extraction
Quantification of indicated bacterial groups in feces of WT-FL and LRP5/6ΔCD11c mice was performed by quantitative PCR as described previously (23, 24). Briefly, fecal pellets were collected from mice and bacterial DNA was extracted with the QIAamp DNA Stool Kit (Qiagen). Quantitative PCR for the 16S rRNA gene was performed with SYBR Green (Bio-Rad). Amounts of indicated bacteria groups were first normalized to that of total bacterial DNA. Abundance of each bacterial group in the feces from WT-FL or LRP5/6ΔCD11c mice was taken as 1 to calculate the relative abundances of corresponding bacterial groups in feces from WT-FL and LRP5/6ΔCD11c mice. Reactions were run with the MyiQ5 ICycler Real-Time PCR Detection System (Bio-Rad). Primers used in this study have been described previously (23, 24).
Histopathology and immunohistochemistry
Sections (5 μm thick) from formalin-fixed and paraffin-embedded colons were placed onto glass slides. H&E-stained sections were blindly scored for severity of colonic inflammation as described previously (23, 24). The degree of inflammation was scored as: 0, no inflammation; 1, mild inflammation or prominent lymphoid aggregates; 2, moderate inflammation; 3, moderate inflammation associated with crypt loss; or 4, severe inflammation with crypt loss and ulceration. Crypt destruction was graded as: 0, no destruction; 1, 1–33% of crypts destroyed; 2, 34–66% of crypts destroyed; or 3, 67–100% of crypts destroyed. The individual scores from inflammation and crypt damage were summed to derive a histological score for colonic inflammation (maximum score of 7).
Measurement of intestinal permeability
Mice were given FITC–dextran by oral gavage at a dose of 0.5 mg/g of body weight. Four hours later, mice were bled and FITC–dextran was quantified in the serum via a fluorescence spectrophotometer.
Myeloperoxidase activity
Pieces of colon (100 mg weight) were homogenized in phosphate buffer (20 mM [pH 7.4]) and centrifuged. The pellet was resuspended in phosphate buffer (50 mM [pH 6]) containing 0.5% hexadecyltrimethylammonium bromide (Sigma-Aldrich). The sample was freeze-thawed and thereafter sonicated, followed by warming to 60°C for 2 h and subsequent centrifugation. Redox reaction of 3,3′,5,5′-tetramethylbenzidine (Sigma-Aldrich) by supernatant was used to determine myeloperoxidase activity. The reaction was terminated with 2N HCl and absorbance was read at 450 nm.
Real-time PCR
Total mRNA was isolated from colon or indicated cell type using the Ω Total RNA Kit according to the manufacturer’s protocol. cDNA was generated using the RNA to cDNA Ecodry Premix Kit (Clontech) according to the manufacturer’s protocol. cDNA was used as a template for quantitative real-time PCR using SYBR Green Master Mix (Bio-Rad) and gene-specific primers (18). PCR analysis was performed using a MyiQ5 ICycler (Bio-Rad). Gene expression was normalized relative to Gapdh.
Western blot of APCs purified from LP
Isolated leukocytes obtained after leukocyte preparation were incubated at 4°C for 30 min with 50 μl each of CD11c microbeads (Miltenyi Biotec). CD11c+ APCs were positively selected from the leukocyte preparation by elution via MACS LS columns (Miltenyi Biotec) and then FACS sorted for CD45+ CD11c+ I-Ab+ cells. The cells were then lysed in radioimmunoprecipitation assay buffer supplemented with protease and phosphatase inhibitors, and the lysates were thereafter used for protein band chemiluminescence detection for nonphosphorylated active β-catenin, total β-catenin, LRP5, LRP6, and β-actin.
Statistical analyses
Statistical analyses were performed using GraphPad Prism software. An unpaired one-tailed Student t test was used to determine statistical significance for mRNA expression levels, Treg percentages and cytokines released by various cell types between different groups. A p value <0.05 (*) was considered to be significant, a p value <0.01 (**) was considered to be very significant, and a p value <0.001 (***) was considered to be extremely significant.
Results
CD11c+ APCs express Wnt ligands in the colon in the steady state and in response to inflammation
We hypothesized that canonical Wnt-mediated signaling in APCs suppresses intestinal inflammation. To better understand the role of canonical Wnt signaling in intestinal homeostasis, we analyzed the expression of Wnt ligands in the colon in the steady state and in response to inflammation. For this, we challenged WT mice with 3.5% DSS, an experimental model of tissue injury and intestinal inflammation. We noted a significant increase in the expression of Wnt2, Wnt3a, Wnt5a, Wnt7a, Wnt8a, Wnt8b, Wnt10a, and Wnt11 in the colon in response to DSS treatment (Supplemental Fig. 1A).
There are two major subsets of APCs in the colonic mucosa: MPs (MHCII+CD11c+CD103−CD11b+CX3CRl+F4/80+CD64+) and DCs (MHCII+CD11c+CX3CR1int/−F4/80−CD64−) (Supplemental Fig. 1B) (27). In addition, DCs can be further subdivided into CD103+ and CD103− subsets (27). Our previous study showed that the canonical Wnt pathway is highly active in intestinal DCs and MPs (25).Thus, we quantified the mRNA expression levels of various Wnt ligands in the DCs and MPs in the colon under steady-state conditions. Colonic CD11c+ DCs and MPs expressed markedly higher levels of Wnt2, Wnt3a, Wnt5a, Wnt7a, Wnt8a, Wnt8b, and Wnt9a compared with splenic DCs (Supplemental Fig. 1C). In addition, colonic CD11c+ MPs appeared to express Wnt ligands to a much greater extent than colonic CD11c+ DCs (Supplemental Fig. 1C). In contrast, colonic epithelial cells expressed markedly lower levels of Wnt ligands compared with colonic DCs and MPs (Supplemental Fig. 1C). Next, we quantified the expression of Wnt ligands in colonic DCs and MPs in response to inflammation. We found that although both cell subsets significantly expressed more Wnt3A, Wnt5B, and Wnt10A during colitis compared with steady state conditions, only MPs substantially increased expression of Wnt2, Wnt8A, Wnt8B, and Wnt11 (Supplemental Fig. 2A, 2B).
Because coreceptors LRP5 and LRP6 are critical for mediating canonical Wnt signaling, we determined whether intestinal APC subsets express these coreceptors. Colonic DCs and MPs isolated from WT mice expressed both LRP5 and LRP6 coreceptors (Supplemental Fig. 2C). In addition, both colonic CD103+ and CD103− DC subsets express LRP5 and LRP6 (Supplemental Fig. 2C). Next, we analyzed the expression of LRP5 and LRP6 in intestinal APCs in response to DSS treatment. We found that both LRP5 and LRP6 are still expressed in both intestinal DCs and MPs, even during inflammatory colitis (Supplemental Fig. 2D).
Deletion of Wnt coreceptors LRP5 and LRP6 in CD11c+ APCs enhances susceptibility to lethal colitis
To address the role of LRP5- and LRP6-mediated signaling in CD11c+ APCs in the intestine, we generated CD11c+ APC–specific deletion of LRP5 (LRP5ΔCD11c) or LRP6 (LRP6ΔCD11c) by crossing LRP5FL mice (LRP5FL/FL) or LRP6FL (LRP6FL/FL) mice (21) with CD11c-cre mice (20). We then challenged LRP5ΔCD11c and LRP6ΔCD11c mice with 3.5% DSS. Both LRP5ΔCD11c and LRP6ΔCD11c mice showed weight loss similar to LRP5FL and LRP6FL mice in response to DSS treatment (Supplemental Fig. 3A, 3B). Because it is possible that LRP5 and LRP6 may play a compensatory role in CD11c+ APCs in response to intestinal inflammation, we then crossed LRP5ΔCD11c and LRP6ΔCD11c mice to generate double-knockout LRP5/6ΔCD11c mice. Western blot analysis of colonic CD11c+ APCs showed depletion of both LRP5 and LRP6 in LRP5/6ΔCD11c mice (Supplemental Fig. 3C). Further analysis of colonic CD11c+ APC subsets showed selective depletion of the coreceptors in both DCs and MPs (Supplemental Fig. 3D). Then, we subjected LRP5/6ΔCD11c and WT-FL (LRP5/6FL/FL) mice to 3.5% DSS treatment and monitored for body weight loss and recovery. LRP5/6ΔCD11c mice were highly susceptible to 3.5% DSS, with increased weight loss beginning on day 2, and failed to recover during the course of treatment (Supplemental Fig. 3E, 3F). In contrast, all WT-FL mice lost less weight compared with LRP5/6ΔCD11c mice and recovered during the course of treatment (Supplemental Fig. 3E, 3F). To study the phenotype of LRP5/6ΔCD11c mice under milder conditions, we repeated the experiment with a lower dose of DSS (2%) treatment. Upon DSS administration at the reduced dose, LRP5/6ΔCD11c mice showed greater weight loss, diarrhea, and rectal bleeding compared with the WT-FL mice (Fig. 1A–C). Moreover, DSS treatment of LRP5/6ΔCD11c mice resulted in a significant reduction in colon length compared with the colons of WT-FL mice (Fig. 1D). Myeloperoxidase activity, a hallmark of colonic inflammation, was markedly increased in the colons of LRP5/6ΔCD11c mice after DSS treatment (Fig. 1E). Histopathological analysis of colons of DSS-treated LRP5/6ΔCD11c mice showed extensive damage to the mucosa, which included epithelial erosion, loss of crypts, and infiltration of immune cells, compared with the colons of DSS-treated WT-FL mice (Fig. 1F, 1G). However, colons from untreated WT-FL and LRP5/6ΔCD11c mice showed no morphological sign of damage or inflammation (data not shown). Consistent with enhanced gut inflammation and delayed recovery, we also observed an increase in FITC–dextran in the serum of DSS-treated LRP5/6ΔCD11c mice after oral gavage (Fig. 1H), indicating severely impaired epithelial barrier integrity. Collectively, our results also show that the absence of both of these coreceptors in CD11c+ APCs in mice results in increased intestinal inflammation with delayed recovery, indicating a possible regulatory role for LRP5/6-mediated signaling in CD11c+ APCs during ongoing intestinal inflammation.
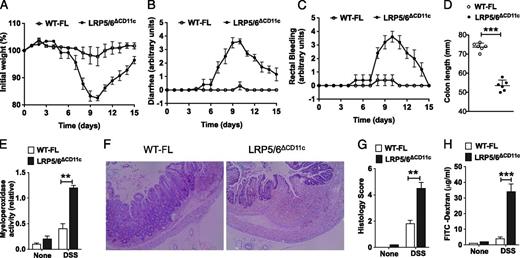
Increased susceptibility of LRP5/6ΔCD11c mice to lethal colitis. WT-FL and LRP5/6ΔCD11c mice were treated with 2% DSS (36–50 kDa) in their drinking water for 7 d before returning to normal water, and at day 10, the colons of mice were analyzed for inflammation. (A–D) Change in body weight, diarrhea, rectal bleeding, and colon length of WT-FL and LRP5/6ΔCD11c mice (n ≥ 5). (E) Myeloperoxidase activity in colon (n ≥ 5). (F) Representative images of H&E-stained colonic sections from DSS-treated WT-FL and LRP5/6ΔCD11c mice (original magnification ×100). (G) Histopathological score (inflammation + epithelial damage) of colons was graded following analysis of H&E-stained cross-sections of colons of DSS-treated WT-FL and LRP5/6ΔCD11c mice. (H) Mice were fed with FITC–dextran on day 10, and 4 h later FITC–dextran was quantified in serum (n = 5). Values are mean ± SEM or representative of at least two independent experiments. **p < 0.01, ***p < 0.001.
Increased susceptibility of LRP5/6ΔCD11c mice to lethal colitis. WT-FL and LRP5/6ΔCD11c mice were treated with 2% DSS (36–50 kDa) in their drinking water for 7 d before returning to normal water, and at day 10, the colons of mice were analyzed for inflammation. (A–D) Change in body weight, diarrhea, rectal bleeding, and colon length of WT-FL and LRP5/6ΔCD11c mice (n ≥ 5). (E) Myeloperoxidase activity in colon (n ≥ 5). (F) Representative images of H&E-stained colonic sections from DSS-treated WT-FL and LRP5/6ΔCD11c mice (original magnification ×100). (G) Histopathological score (inflammation + epithelial damage) of colons was graded following analysis of H&E-stained cross-sections of colons of DSS-treated WT-FL and LRP5/6ΔCD11c mice. (H) Mice were fed with FITC–dextran on day 10, and 4 h later FITC–dextran was quantified in serum (n = 5). Values are mean ± SEM or representative of at least two independent experiments. **p < 0.01, ***p < 0.001.
Absence of LRP5/6 signaling in intestinal APCs fosters inflammatory microenvironment in the colon
In the DSS model of intestinal inflammation, inflammatory cytokines produced by innate immune cells present in the gut microenvironment promote colitis and augment tissue injury (28). Thus, we analyzed the expression of immune-regulatory and inflammatory factors that suppress or promote colonic inflammation. Colons of DSS-treated LRP5/6ΔCD11c mice expressed higher levels of inflammatory cytokines such as IL-1β, IL-6, TNF-α, IFN-γ, and IL-17 compared with colons of DSS-treated WT-FL mice (Fig. 2A). In contrast, the colons of DSS-treated LRP5/6ΔCD11c mice expressed lower levels of IL-10 and IL-22 compared with those of colons of WT-FL mice (Fig. 2A). Consistent with these observations, colon explant cultures showed that colons of DSS-treated LRP5/6ΔCD11c mice produced higher levels of inflammatory cytokines and lower levels of IL-10 and IL-22 compared with colons of WT-FL mice (Fig. 2B). Taken together, these findings demonstrate an imbalance in the production of anti-inflammatory molecules versus inflammatory molecules that favors increased susceptibility of LRP5/6ΔCD11c mice to DSS-induced colitis and inflammation-associated tissue injury.

LRP5/6 signaling in CD11c+ APCs suppresses the expression of inflammatory factors in the colon. (A) RNA was extracted from colons of untreated and DSS-treated WT-FL and LRP5/6ΔCD11c mice. Expression of indicated genes was quantified by quantitative PCR. (B) Excised colon samples of untreated and DSS-treated WT-FL and LRP5/6ΔCD11c mice were cultured for 2 d ex vivo, and the cytokine levels in the culture supernatants were quantified by ELISA. The error bars indicate mean ± SEM of five to six mice per group or representative of at least two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
LRP5/6 signaling in CD11c+ APCs suppresses the expression of inflammatory factors in the colon. (A) RNA was extracted from colons of untreated and DSS-treated WT-FL and LRP5/6ΔCD11c mice. Expression of indicated genes was quantified by quantitative PCR. (B) Excised colon samples of untreated and DSS-treated WT-FL and LRP5/6ΔCD11c mice were cultured for 2 d ex vivo, and the cytokine levels in the culture supernatants were quantified by ELISA. The error bars indicate mean ± SEM of five to six mice per group or representative of at least two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
LRP5/6 signaling in colonic CD11c+ APCs regulates the balance between Treg and effector T cell frequency in the colon in a microbiota-dependent manner
The balance between regulatory and effector T cells is critical for gut homeostasis (29–31). Intestinal DCs and MPs play a critical role in maintaining balance between regulatory and effector T cells (7, 9, 32, 33). Therefore, based on the above results, we sought to determine whether CD11c+ APC-intrinsic LRP5/6 signaling is critical for T cell homeostasis. As Th1/Th17 cells in the colon promote inflammation (29), we quantified the frequency of Th1/Th17 cells in the intestine of LRP5/6ΔCD11c and WT-FL mice under steady-state conditions. Remarkably, LRP5/6ΔCD11c mice displayed higher frequencies of CD4+ cells producing IFN-γ or IL-17A in the colon compared with WT mice (Fig. 3A, 3B, top panels showing mice without antibiotic treatment [−Abx]). In contrast, we observed markedly reduced levels of IL-10-producing Tr1 cells and Foxp3+ Tregs in the colon of LRP5/6ΔCD11c mice compared with that of WT-FL mice (Fig. 3C, 3D). These observations were further associated with a significant increase in IFN-γ and IL-17A and a marked reduction in IL-10 in the colons of LRP5/6ΔCD11c mice as compared with WT mice (Fig. 3E). Interestingly, there was no significant difference in the frequencies of Th1 and Th17 cells in the spleens of LRP5/6ΔCD11c mice versus WΤ-FL mice (data not shown). These observations suggest that an increase in Th1 and Th17 cells in LRP5/6ΔCD11c mice is specific to the colon.

LRP5/6 signaling in intestinal APCs limits inflammatory responses to commensal flora. (A–D) FACS plots representing percentages or cumulative frequencies of CD4+ T cells positive for IL-17A/IFN-γ or Foxp3 isolated from colons of WT-FL and LRP5/6ΔCD11c mice treated with (+Abx, bottom panels) or without (−Abx, top panels) antibiotics (n = 8). (E) Excised colon samples in panel A were cultured for 2 d ex vivo, and then the secreted IL-17A, IFN-γ, and IL-10 amounts in the culture supernatants were quantified by ELISA (n = 5). (F) Relative quantification of different bacterial species in the fecal material from LRP5/6ΔCD11c mice compared with WT-FL mice as analyzed by quantitative PCR analysis (n > 5). Values are mean ± SEM or representative of at least three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
LRP5/6 signaling in intestinal APCs limits inflammatory responses to commensal flora. (A–D) FACS plots representing percentages or cumulative frequencies of CD4+ T cells positive for IL-17A/IFN-γ or Foxp3 isolated from colons of WT-FL and LRP5/6ΔCD11c mice treated with (+Abx, bottom panels) or without (−Abx, top panels) antibiotics (n = 8). (E) Excised colon samples in panel A were cultured for 2 d ex vivo, and then the secreted IL-17A, IFN-γ, and IL-10 amounts in the culture supernatants were quantified by ELISA (n = 5). (F) Relative quantification of different bacterial species in the fecal material from LRP5/6ΔCD11c mice compared with WT-FL mice as analyzed by quantitative PCR analysis (n > 5). Values are mean ± SEM or representative of at least three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
The intestinal microbiota has been observed to play an important role in the induction of CD4+ effector T cells and Tregs (5, 30). Therefore, we next assessed whether increased Th17/Th1 cell frequencies and inflammatory cytokine expression in the colon of LRP5/6ΔCD11c mice is due to gut microflora by performing microbiota depletion studies using an antibiotic mixture (25). Antibiotic treatment resulted in a marked reduction in the frequencies of Th17 and Th1 cells in the colon of LRP5/6ΔCD11c mice as compared with untreated control mice (Fig. 3A, 3B). A similar change in frequency was not observed, however, for IL-10–producing Tr1 or Foxp3-expressing Tregs (Fig. 3C, 3D). Consistent with these observations, colons of antibiotic-treated LRP5/6ΔCD11c mice produced significantly less IL-17A and IFN-γ, but not IL-10, compared with control −Abx colons (Fig. 3E). Collectively, these findings suggest an important role for LRP5/6 in CD11c+ APCs regulating a delicate balance between Treg and effector T cell numbers in the colon.
Because depleting microbiota reduced both Th17/Th1 cells and inflammatory cytokine levels in the colons of LRP5/6ΔCD11c mice, we next investigated whether LRP5/6ΔCD11c mice harbor altered commensal microflora. For this, we quantified the relative levels of different bacterial species in the feces of LRP5/6ΔCD11c mice and WT mice. Interestingly, we observed an increased presence of segmented filamentous bacteria (SFB), Prevotellaceae, and TM7 groups of commensal bacteria in LRP5/6ΔCD11c mice as compared with WT-FL mice (Fig. 3F). In contrast, we observed markedly reduced levels of Clostridiales and Bacteroides groups of commensal bacteria in LRP5/6ΔCD11c mice (Fig. 3F). This result provides evidence for altered microflora in the intestine of LRP5/6ΔCD11c mice and explains the enhanced susceptibility of LRP5/6ΔCD11c mice to colonic inflammation.
LRP5/6 signaling imparts regulatory phenotype on colonic APCs
Intestinal APCs express immune-regulatory factors such as IDO, IL-10, and retinoic acid (RA) that preferentially drive Treg responses while limiting Th1/Th17 responses (7, 9, 32, 33). We reasoned that decreased frequencies of Tregs and increased frequencies of Th1 and Th17 cells in the colons of LRP5/6ΔCD11c mice are due to the loss of an immune-regulatory phenotype of colonic DCs and/or MPs. Thus, we analyzed the expression levels of inflammatory and anti-inflammatory factors in colonic DCs and MPs of LRP5/6ΔCD11c mice. Colonic DCs and MPs isolated from the LRP5/6ΔCD11c mice expressed markedly lower mRNA levels of immune-regulatory factors such as Aldh1a1, Aldh1a2, IL-10, and IDO1 (Fig. 4A). In contrast, LRP5/6-deficient colonic DCs and MPs expressed significantly higher levels of inflammatory factors such as IL-1β and IL-6 (Fig. 4B). Consistent with these observations, colonic DCs and MPs from LRP5/6ΔCD11c mice cultured ex vivo produced higher levels of cytokines such as IL-1β and IL-6 and lower levels of IL-10 compared with colonic DCs and MPs from WT-FL mice (Fig. 4C).

LRP5/6 signaling imparts anti-inflammatory phenotype on colonic DCs and MPs. (A and B) Quantitative real-time PCR analysis of the mRNA of indicated genes in colonic CD11c+ DCs and MPs sorted from WT-FL and LRP5/6ΔCD11c mice. Data are presented as fold change relative to WT (n = 5). (C) Sorted colonic CD11c+ DCs and MPs from WT-FL and LRP5/6ΔCD11c mice were cultured for 2 d ex vivo, and IL-1β, IL-6, and IL-10 cytokine amounts in the culture supernatants were quantified by ELISA (n = 4). (D and E) Representative FACS plot (D) and cumulative frequencies (E) of adoptively transferred naive OT-II CD4+ T cells positive for IFN-γ, IL-17A, IL-10, and Foxp3 isolated from colons of WT-FL and LRP5/6ΔCD11c mice treated orally with OVA protein (n = 8). Values are mean ± SEM or representative of at least three independent experiments. **p < 0.01, ***p < 0.001.
LRP5/6 signaling imparts anti-inflammatory phenotype on colonic DCs and MPs. (A and B) Quantitative real-time PCR analysis of the mRNA of indicated genes in colonic CD11c+ DCs and MPs sorted from WT-FL and LRP5/6ΔCD11c mice. Data are presented as fold change relative to WT (n = 5). (C) Sorted colonic CD11c+ DCs and MPs from WT-FL and LRP5/6ΔCD11c mice were cultured for 2 d ex vivo, and IL-1β, IL-6, and IL-10 cytokine amounts in the culture supernatants were quantified by ELISA (n = 4). (D and E) Representative FACS plot (D) and cumulative frequencies (E) of adoptively transferred naive OT-II CD4+ T cells positive for IFN-γ, IL-17A, IL-10, and Foxp3 isolated from colons of WT-FL and LRP5/6ΔCD11c mice treated orally with OVA protein (n = 8). Values are mean ± SEM or representative of at least three independent experiments. **p < 0.01, ***p < 0.001.
The type of cytokine milieu present in the gut microenvironment drives the differentiation and expansion of effector T cells and Tregs (29, 30). Thus, we investigated the in vivo role of LRP5/6 signaling in intestinal APCs in the induction of Treg/Th1/Th17 cells in response to oral Ag using an Ag-specific CD4+ T cell transfer system. We adoptively transferred naive OT-II Thy1.1+ cells into LRP5/6ΔCD11c and WT-FL mice and fed them with OVA, the cognate Ag for OT-II cells. Intracellular cytokine analysis on day 6 posttransfer showed a significant increase in naive OT-II Thy1.1 CD4+ T cell differentiation toward Th1 and Th17 cells in LRP5/6ΔCD11c mice compared with WT mice in the colon (Fig. 4D, 4E). Further characterization of transferred OT-II T cells showed a marked decrease in the differentiation of Foxp3+ Tregs and IL-10–producing Tr1 cells in the colon of LRP5/6ΔCD11c mice compared with WT mice (Fig. 4D, 4E). Thus, these data demonstrate that LRP5/6 signaling imparts an anti-inflammatory phenotype on intestinal APCs by inducing the expression of key immune-regulatory genes while suppressing the expression of inflammatory cytokines.
Activation of β-catenin in APCs ameliorates intestinal inflammation in LRP5/6ΔCD11c mice
The transcriptional cofactor β-catenin is a key downstream mediator of canonical Wnt signaling (3, 4). Previous studies have shown that the LRP5/6-mediated signaling activates β-catenin in splenic DCs and that β-catenin is highly active in intestinal APCs (18, 25). Deletion of β-catenin in CD11c+ APCs renders mice susceptible to DSS-induced colitis (25). Thus, we asked whether LRP5/6 signaling is critical for β-catenin activation in intestinal APCs. We quantified the β-catenin activation status by immunoblot using Abs that specifically detect the active form of β-catenin. Colonic CD11c+ APCs deficient in LRP5/6 showed markedly reduced levels of the active form of β-catenin compared with WT-FL colonic CD11c+ APCs (Fig. 5A). However, both WT and LRP5/6-deficient colonic CD11c+ APCs showed comparable levels of total β-catenin and β-actin. These observations suggest that LRP5/6 signaling is critical for the activation of β-catenin in intestinal APCs.
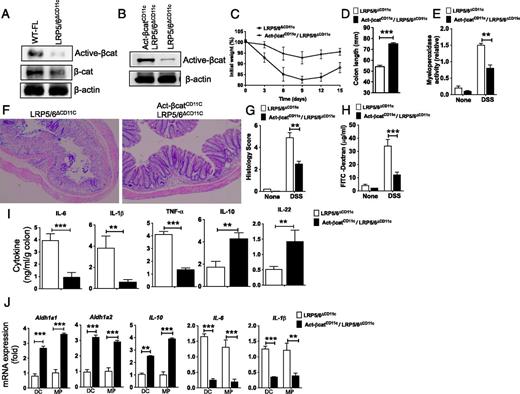
Colitis is remediated in LRP5/6ΔCD11c mice following activation of β-catenin in CD11c+ APCs. (A) Western blot of CD11c+ APCs isolated from the colon, showing differences in relative expression of active and total β-catenin protein between WT-FL and LRP5/6ΔCD11c mice. (B) Western blot of CD11c+ APCs from the colonic LP, showing differences in relative expression of active β-catenin protein between LRP5/6ΔCD11c and LRP5/6ΔCD11c X Act-βcatCD11c mice. (C and D) Weight loss and colon lengths of LRP5/6ΔCD11c and Act-βcatCD11c/LRP5/6ΔCD11c mice subjected to 2% DSS (n > 5). (E) Myeloperoxidase activity in colon (n ≥ 5). (F) Representative images of H&E-stained colonic sections from LRP5/6ΔAPC and Act-βcatAPC X LRP5/6ΔAPC mice (original magnification ×100). (G) Histopathological score (inflammation + epithelial damage) of colons was graded following analysis of H&E-stained cross-sections of colons of DSS-treated LRP5/6ΔAPC and Act-βcatAPC X LRP5/6ΔAPC mice. (H) Mice were fed with FITC–dextran on day 15, and 4 h later FITC–dextran was quantified in serum (n = 5). (I) Sorted colonic DCs and MPs from LRP5/6ΔCD11c and Act-βcatCD11c X LRP5/6ΔCD11c mice were cultured for 2 d ex vivo, and IL-1β, IL-6, TNF-α, IL-22, and IL-10 cytokine amounts in the culture supernatants were quantified by ELISA (n = 3). (J) Quantitative real-time PCR analysis of the mRNA of indicated genes relative to Gapdh in colonic DCs and MPs sorted from LRP5/6ΔCD11c and Act-βcatCD11c X LRP5/6ΔCD11c mice (n = 4). Values are mean ± SEM or representative of at least two independent experiments. **p < 0.01, ***p < 0.001.
Colitis is remediated in LRP5/6ΔCD11c mice following activation of β-catenin in CD11c+ APCs. (A) Western blot of CD11c+ APCs isolated from the colon, showing differences in relative expression of active and total β-catenin protein between WT-FL and LRP5/6ΔCD11c mice. (B) Western blot of CD11c+ APCs from the colonic LP, showing differences in relative expression of active β-catenin protein between LRP5/6ΔCD11c and LRP5/6ΔCD11c X Act-βcatCD11c mice. (C and D) Weight loss and colon lengths of LRP5/6ΔCD11c and Act-βcatCD11c/LRP5/6ΔCD11c mice subjected to 2% DSS (n > 5). (E) Myeloperoxidase activity in colon (n ≥ 5). (F) Representative images of H&E-stained colonic sections from LRP5/6ΔAPC and Act-βcatAPC X LRP5/6ΔAPC mice (original magnification ×100). (G) Histopathological score (inflammation + epithelial damage) of colons was graded following analysis of H&E-stained cross-sections of colons of DSS-treated LRP5/6ΔAPC and Act-βcatAPC X LRP5/6ΔAPC mice. (H) Mice were fed with FITC–dextran on day 15, and 4 h later FITC–dextran was quantified in serum (n = 5). (I) Sorted colonic DCs and MPs from LRP5/6ΔCD11c and Act-βcatCD11c X LRP5/6ΔCD11c mice were cultured for 2 d ex vivo, and IL-1β, IL-6, TNF-α, IL-22, and IL-10 cytokine amounts in the culture supernatants were quantified by ELISA (n = 3). (J) Quantitative real-time PCR analysis of the mRNA of indicated genes relative to Gapdh in colonic DCs and MPs sorted from LRP5/6ΔCD11c and Act-βcatCD11c X LRP5/6ΔCD11c mice (n = 4). Values are mean ± SEM or representative of at least two independent experiments. **p < 0.01, ***p < 0.001.
Next, we asked whether activation of the β-catenin pathway in LRP5/6-deficient APCs could ameliorate intestinal inflammation in LRP5/6ΔCD11c mice. To test this, we crossed mice expressing an active form of the β-catenin in CD11c+ APCs (β-cat+/lox (ex3)/CD11c-cre+, referred to as Act-βcatCD11C after this) onto an LRP5/6ΔCD11c background. Immunoblot analysis of colonic CD11c+ APCs showed a marked increase in the expression of the active form of β-catenin in Act-βcatCD11c/LRP5/6ΔCD11c mice compared with that of LRP5/6ΔCD11c mice (Fig. 5B). We then subjected LRP5/6ΔCD11c and Act-βcatCD11c/LRP5/6ΔCD11c mice to DSS treatment. Upon DSS administration, Act-βcatCD11c/LRP5/6ΔCD11c mice showed a marked decrease in colitis severity compared with LRP5/6ΔCD11c mice, which included lesser weight loss, inflammation, colon shortening, and myeloperoxidase activity (Fig. 5C–E). Histopathology of colons of DSS-treated Act-βcatCD11c/LRP5/6ΔCD11c mice showed less extensive damage to the mucosa regarding epithelial erosion, loss of crypts, and infiltration of immune cells compared with the colons of DSS-treated of LRP5/6ΔCD11c mice (Fig. 5F, 5G). In addition, we also observed a decrease in FITC–dextran in the serum of DSS-treated Act-βcatCD11c/LRP5/6ΔCD11c mice (Fig. 5H), indicating less damage to the integrity of the epithelial barrier. Consistent with diminished gut inflammation, colon explant cultures showed that colons of DSS-treated Act-βcatCD11c/LRP5/6ΔCD11c mice produced lower levels of inflammatory cytokines (IL-1β, IL-6, TNF-α) and higher levels of IL-10 and IL-22 compared with colons of LRP5/6ΔCD11c mice (Fig. 5I). Next, we analyzed the expression levels of inflammatory and anti-inflammatory factors in colonic CD11c+ DCs and MPs of Act-βcatCD11c/LRP5/6ΔCD11c mice. Colonic DCs and MPs isolated from the Act-βcatCD11c/LRP5/6ΔCD11c mice expressed markedly higher mRNA levels of Aldh1a1, Aldh1a2, and IL-10 and markedly lower levels of IL-6 and IL-1β (Fig. 5J). Collectively, these observations suggest that β-catenin is a key downstream mediator of LRP5/6 signaling in intestinal APCs and that activation of β-catenin in LRP5/6ΔCD11c mice ameliorates DSS-induced colitis and inflammation-associated tissue injury.
Discussion
The current study defines an essential role for the canonical Wnt-signaling pathway in CD11c+ APCs in controlling colonic inflammation to the microbiota. Accordingly, LRP5/6ΔCD11c led to an increased expression of IL-1β, IL-6, and TNF-α, with diminished production of IL-10 and RA. Consequently, the absence of LRP5/6-mediated signaling in colonic CD11c+ APCs resulted in a loss of T cell homeostasis in the colon because of altered gut microbiota. Furthermore, the current study shows β-catenin as a key downstream mediator of LRP5/6 signaling in intestinal APCs. Finally, in LRP5/6ΔCD11c mice, conditional activation of β-catenin in CD11c+ APCs also results in reduced intestinal inflammation. Collectively, these findings support the hypothesis that canonical Wnt signaling imparts an anti-inflammatory phenotype on colonic APCs and is critical for suppressing colonic inflammation to commensal flora.
The intestine contains high levels of Wnt ligands, and the Wnt-signaling pathway is critical for gut development and homeostasis (1, 2). However, the focus of most research has been on the effects of this pathway on intestinal stem cell proliferation as well as its effects on cancer initiation and progression. Wnt ligands in the gut environment can also act on immune cells and shape mucosal immune responses. An unanswered question, however, is whether canonical Wnt signaling in APCs is critical for suppressing inflammation in the gut. The present study shows that genetic deletion of LRP5/6 specifically in CD11c+ APCs in mice results in enhanced intestinal inflammation in response to DSS treatment. In APCs, Wnts activate both β-catenin–dependent and β-catenin–independent signaling pathways (4). Conditioning of DCs with Wnt ligands activates the β-catenin pathway (15, 18) and programs them to a regulatory state (18, 34, 35). The β-catenin pathway is highly active in intestinal APCs and is critical for mediating immune tolerance to gut microflora (25). Mice deficient in β-catenin specifically in CD11c+ APCs are highly susceptible to DSS-induced colitis and other inflammatory diseases (18, 25, 36). In addition to Wnts, multiple signaling pathways activate β-catenin (36, 37). β-catenin activation is markedly reduced in intestinal CD11c+ APCs deficient in LRP5/6. Further, our study shows that, conditional expression of the active form of β-catenin in CD11c+ APCs in LRP5/6ΔCD11c mice suppresses colonic inflammation and markedly reduces inflammation-mediated tissue injury. DCs and MPs are also present in inflamed tissues, where they control inflammation and express factors that are critical for tissue repair. Several studies have reported that Wnt ligands play a key role in wound healing and intestinal repair (38–40). In line with these studies, we also observe a marked increase in the expression of Wnt ligands upon inflammation in the colon. The present study also shows that CD11c+ APCs isolated from the inflamed colon express coreceptors LRP5 and LRP6 and are the major producers of Wnt ligands in the inflamed tissues. In settings of chronic inflammatory diseases, canonical Wnt signaling in DCs acts as a counter-regulatory mechanism to control inflammation and restore homeostasis (16–19, 41). These observations suggest that LRP5/6-dependent signaling in intestinal APCs plays a similar role in mediating immune tolerance in response to commensal flora.
A delicate balance between Tregs versus pathological effector T cells underlies disease progression in many inflammatory diseases, including IBD (7, 9, 32, 33). Intestinal DCs and MPs play a critical role in controlling this balance between tolerance and inflammation (7, 9, 32, 33). Intestinal DCs and MPs control intestinal inflammation by expressing immune-regulatory factors such as RA, IL-10, and IDO (7, 9, 32, 33). Studies conducted with transplantable tumors in mice and classical murine melanoma models have shown that Wnt ligands in the tumor microenvironment suppress host immune responses by inducing RA, IL-10, and IDO in DCs (12–15). The present study shows that canonical Wnt signaling in intestinal APCs is critical for the induction of these immune-regulatory factors and in imparting anti-inflammatory properties while limiting the expression of inflammatory cytokines. These regulatory factors are critical for driving Treg differentiation and expansion while limiting the differentiation of Th1/Th17 cells in the gut (7, 9, 32, 33). Deletion of LRP5/6 in CD11c+ APCs resulted in the loss of T cell homeostasis with reduced numbers of Tregs and increased numbers of Th1/Th17 cells in the colon. This imbalance in T cell subsets was due to increased expression of inflammatory cytokines by colonic APCs that drive Th1/Th17 cell differentiation. This is dependent on the commensal flora, as antibiotic mixture treatment markedly reduced the expression of inflammatory cytokines such as IL-17A or IFN-γ, resulting in reduced inflammation and damage accrued via pathological effector T cells. Microbiota-induced IL-1β and IL-6 are critical for the development of intestinal Th-17 cells, and the gut-resident APCs are the main source of IL-1β and IL-6 (42). Because colonic APCs express LRP5/6, it is possible that the canonical Wnt-signaling pathway might play a critical role in regulating IL-1β and other inflammatory cytokines in intestinal APCs in response to microbes under steady-state conditions and inflammatory conditions. These observations suggest that LRP5/6-dependent signaling in intestinal APCs plays a key role in mediating immune tolerance in response to commensal flora.
Genetic modification of the host leads to microbial dysbiosis resulting in host susceptibility to colonic inflammation (23, 24, 43–45). The gut microbiota plays an important role in shaping mucosal innate and adaptive immune responses (7, 8). Further analysis of microbial species in the stool of LRP5/6ΔCD11c mice revealed an increased representation of SFB, Prevotellaceae, and TM7 groups of commensal bacteria. In line with these observations, past studies have shown that increased representation of SFB, Prevotellaceae, and TM7 groups of commensal bacteria is associated with enhanced risk of colitis in mice (23, 24, 43, 45). Although LRP5/6ΔCD11c mice harbor altered microflora, the colons of these mice showed no morphological signs of damage up to 16 wk of age.
IL-22 is critical for maintaining the integrity of epithelial cell barrier, mucous production, and epithelial cell regeneration and repair (46). IL-22 regulates the expression of antimicrobial peptides such as RegIIIβ, RegIIIγ, S100A8, and S100A9 by intestinal epithelial cells, which is critical for barrier immunity (47–49). Past studies have shown protective effects of IL-22 in the DSS-induced colitis model (50), T cell transfer model of IBD (50, 51), and Th2-driven UC model (52). In mice, IL-22 deficiency is associated with commensal dysbiosis and aberrant expansion of SFB, Prevotellaceae, and TM7 groups of commensal bacteria (24, 45, 53). Consistent with these observations, deletion of LRP5/6 in CD11c+ APCs resulted in markedly lower levels of IL-22 both under homeostatic conditions and under inflammatory conditions. In addition, LRP5/6ΔCD11c mice expressed low levels of antimicrobial peptides RegIIIβ and RegIIIγ and calprotectins (S100A8/S100A9) in the colon (Supplemental Fig. 4). In contrast, activation of β-catenin in LRP5/6ΔCD11c mice resulted in a significant increase in IL-22 in the colon. An important unresolved question is how the LRP5/6 signaling in CD11c+ APCs regulates IL-22 expression in the colon. Past studies have shown that RA and IL-10 produced by intestinal APCs can exert paracrine effects on innate lymphoid cells and epithelial cells to augment intestinal barrier immunity against gut flora (54–58).
In summary, the current study shows that the loss of canonical Wnt signaling in CD11c+ intestinal APCs, represented by a CD11c-specific knockout of the LRP5/6 coreceptors, results in the exacerbation of intestinal inflammation in a murine model of UC. This was associated with loss of regulatory phenotypes in intestinal APCs, which expressed lower levels of immune-regulatory factors such as RA and IL-10. A major consequence of these altered characteristics in LRP5/6ΔCD11c mice was due to a loss of T cell homeostasis as a result of an altered gut microbiota. In addition, the current study indicates that the canonical Wnt-signaling pathway plays a key role in regulating commensal homeostasis by regulating the expression of IL-22 and antimicrobial peptides in the gut. Additionally, the exacerbation of intestinal inflammation appeared to be largely due to aberrant downstream β-catenin signaling. These results ultimately portray a significant role for canonical Wnt signaling in intestinal APCs in regulating intestinal inflammation to the commensal microbiota as a result of establishing regulatory phenotypes in both CD11c+ intestinal APCs and T cells. Whether the microbiota plays an important role in regulating the canonical Wnt-signaling pathway in the steady state to mediate immune homeostasis remains largely to be elucidated. Furthermore, how this intestinal APC-intrinsic signaling may be involved in inflammation-associated colon carcinogenesis and other disease states associated with a loss in immune homeostasis in the gut remains unknown. Still, these results show that targeting the canonical Wnt-signaling pathway in intestinal APC subsets may provide a promising treatment strategy for the remediation of colitis and other autoimmune disorders.
Acknowledgements
We thank Dr. Brat O. Williams (Van Andel Research Institute, Grand Rapids, MI) and Dr. Makoto Taketo (Kyoto University Graduate School of Medicine, Kyoto, Japan) for kindly providing LRP5FL, LRP6FL, and β-Catflox(ex3) mice. We thank Jeanene Pihkala and Ningchun Xu for technical help with FACS sorting and analysis, Janice Randall for help with mouse husbandry, and our colleagues in the Augusta University Georgia Cancer Center for constructive comments on various aspects of this study.
Footnotes
This work was supported by National Institutes of Health Award DK097271 (to S.M.).
The online version of this article contains supplemental material.
Abbreviations used in this article:
- −Abx
without antibiotic treatment
- DC
dendritic cell
- DSS
dextran sodium sulfate
- IBD
inflammatory bowel disease
- LP
lamina propria
- LRP5/6
low-density lipoprotein receptor–related proteins 5 and 6
- MP
macrophage
- Treg
regulatory T cell
- UC
ulcerative colitis
- WT
wild-type
- WT-FL
wild-type floxed
- RA
retinoic acid
- SFB
segmented filamentous bacteria.
References
Disclosures
The authors have no financial conflicts of interest.
