

Abstract
Group A streptococcal infections are a significant cause of global morbidity and mortality. A leading vaccine candidate is the surface M protein, a major virulence determinant and protective Ag. One obstacle to the development of M protein–based vaccines is the >200 different M types defined by the N-terminal sequences that contain protective epitopes. Despite sequence variability, M proteins share coiled-coil structural motifs that bind host proteins required for virulence. In this study, we exploit this potential Achilles heel of conserved structure to predict cross-reactive M peptides that could serve as broadly protective vaccine Ags. Combining sequences with structural predictions, six heterologous M peptides in a sequence-related cluster were predicted to elicit cross-reactive Abs with the remaining five nonvaccine M types in the cluster. The six-valent vaccine elicited Abs in rabbits that reacted with all 11 M peptides in the cluster and functional opsonic Abs against vaccine and nonvaccine M types in the cluster. We next immunized mice with four sequence-unrelated M peptides predicted to contain different coiled-coil propensities and tested the antisera for cross-reactivity against 41 heterologous M peptides. Based on these results, we developed an improved algorithm to select cross-reactive peptide pairs using additional parameters of coiled-coil length and propensity. The revised algorithm accurately predicted cross-reactive Ab binding, improving the Matthews correlation coefficient from 0.42 to 0.74. These results form the basis for selecting the minimum number of N-terminal M peptides to include in potentially broadly efficacious multivalent vaccines that could impact the overall global burden of group A streptococcal diseases.
Introduction
Group A streptococcus (Strep A) is responsible for ∼618 million infections and 500,000 deaths yearly (1). The M protein of Strep A is a major determinant of virulence and serves as a protective Ag (2–7). The M protein emanates from the bacteria as a mostly α-helical coiled-coil dimer with its hypervariable N terminus exposed on the surface and its more conserved C terminus anchored in the cell wall (Fig. 1A) (4, 8). The serologic types of Strep A, of which there are more than 200, are defined by the hypervariable N-terminal 50 aa residues of the M protein (9).
Previous studies have demonstrated that Abs against N-terminal M peptides promote opsonization (10) and protect animals from challenge infections (11, 12). Historically, protective immunity was considered M type–specific (13, 14), but more recent studies have shown that the M proteins share sequence similarities, the M proteins can be grouped into sequence-based clusters, and Abs against one N-terminal M peptide may cross-react with others in the same cluster (15–17). Additional evidence indicates that variability within the N-terminal sequence of the M protein is constrained by structural requirements for the binding of host proteins that enhance virulence, such as the complement regulating proteins C4 binding protein (C4BP) and factor H (18–20).
The goal of the current study was to take advantage of the fact that structure is generally more highly conserved than sequence. We developed an algorithm, based on both sequence and structural similarity, that can accurately predict which M peptides are most likely to elicit cross-protective Abs when incorporated into multivalent vaccines, thus improving vaccine efficacy against a significant percentage of epidemiologically important M types of Strep A. Our strategy for vaccine design focuses on the N-terminal 50 residues of the mature M proteins (Fig. 1B). This region contains epitopes that elicit Abs with the greatest opsonic (protective) potential (21, 22) and are least likely to elicit potentially harmful Abs that may cross-react with human tissues (10, 12, 23, 24). We previously constructed a phylogenetic sequence-based tree of 117 N-terminal (50 aa) M peptides (17) from epidemiologically important M types of Strep A and divided them into seven sequence-related clusters (N-terminal clusters [NTC]) based on loosely rooted branches (Fig. 1C). In a recent study, we focused on one sequence-based cluster containing 21 M types (NTC6), using a combination of sequence identity, Ab binding, and cheminformatics to select six vaccine peptides that were predicted to cross-react with all 15 nonvaccine M peptides (17). The vaccine antisera cross-reacted with 10 of the 15 nonvaccine peptides. Interestingly, two of the non–cross-reactive peptides shared 50% or greater sequence identity with the vaccine peptides. Retrospective structural analysis revealed that significant sequence identity at corresponding polar amino acid sites within the coiled-coil α-helical heptad repeats between vaccine and nonvaccine peptides accurately distinguished cross-reactive from non–cross-reactive peptides. We subsequently developed a scoring algorithm (17) based on the sequence identity at polar heptad sites. In the current study, we improve significantly upon the previous algorithm. We followed a sequential process starting with assessments of sequence and structural characteristics, selection of vaccine peptides from a different N-terminal M peptide cluster using the original scoring algorithm, design and production of a multivalent M peptide vaccine, immunization of animals, and then validation of the predicted outcome based on functional immunoassays. We next extended these observations by immunizing groups of mice with four different M peptides having different structural characteristics and testing the immune sera for cross-reactivity against a comprehensive panel of heterologous peptides. Based on these results, we developed a refined algorithm that not only considered stereochemical similarity between the predicted solvent-exposed sites of an M peptide pair but also included other structural parameters such as predicted coiled-coil lengths and coiled-coil propensities of M peptides. The results demonstrate that cross-reactivity predictions among M peptides are improved significantly by including these additional parameters. Finally, we applied the algorithm to the entirety of globally prevalent group A streptococcal M types to predict the selection of vaccine M peptides needed to construct a potentially broadly efficacious Strep A vaccine that could be deployed in all geographic locations.
Materials and Methods
Clustering of M types based on their N-terminal 50-residue sequences
The 117 epidemiologically important M types were analyzed for N-terminal 50-mer sequence identities, and a phylogenetic tree of these M types was generated with Geneious 11.1 (https://www.geneious.com) using neighbor joining and bootstrapping. The tree resulted in seven NTCs (17).
Coiled-coil domain predictions within N-terminal M peptides
The MARCOIL program (25) (hidden Markov model training based on 9FAM, transition matrix: MARCOIL-H) was used to predict the coiled-coil subsequence within the N-terminal 50-mer M peptides and their corresponding heptad registers. This program calculates the probability of each of the residues of a given sequence being in a coiled-coil state as well its most likely heptad site. It thus enables an overview of the predicted coiled-coil probability for the entire sequence. We validated MARCOIL by comparing heptad assignments of the available M protein crystal structures as detected by SOCKET (26) with the heptad assignment predicted by MARCOIL and observed that in each case the actual heptad assignment and the predicted heptad assignment were identical. Because the M protein crystal structures were deposited in the Protein Data Bank after MARCOIL was developed, there is no overlap of our testing dataset with the MARCOIL training data, and we were thus able to obtain an independent and satisfactory evaluation of the program’s utility. Additionally, a comparative analysis of MARCOIL with three other coiled-coil prediction programs, Multicoil2, Paircoil2, and Ncoils, run against all NTC5 sequences revealed a 100% match in the heptad registers. Any residue within the 50-mer with a MARCOIL score >0.02 was considered a part of the coiled-coil domain or coiled-coil subsequence. We chose 0.02 as the threshold as we observed that residues in the M protein sequences that had a score of at least 0.02 as predicted by MARCOIL were resolved by x-ray crystallography as coiled-coils (17).
Two-tiered approach to predict Ab cross-reactivity
To evaluate the cross-reactive potential between any two N-terminal M peptides, we developed a two-tiered approach. First, we determined which pairs of M peptides shared significant sequence identity (>40%) within their 50-mers or within their coiled-coil domains. Second, we computed the empirical heptad similarity score that characterizes the degree of sequence homology/identity between the corresponding predicted solvent-exposed heptad sites of two coiled-coil N-terminal peptides. M peptide pairs with shared sequence identity of >40% and heptad similarity score ≥10.5 were predicted to contain cross-reactive epitopes.
Step a. Sequence identity calculation between pairs of N-terminal peptides
Pairwise global sequence alignments were performed using the Needleman–Wunsch alignment (27), and a threshold of >40% was used to distinguish pairs with cross-reactive potential from pairs with no cross-reactive potential.
Step b. Heptad score calculation between pair of N-terminal peptides
Pairwise global sequence alignment between sequences at corresponding polar heptad sites of two coiled-coil M peptides was also performed using the Needleman–Wunsch alignment.
The heptad score HMx,My between a pair of N-terminal coiled-coil M peptides Mx and My is calculated as follows:
where HMx,My represents the heptad score between two N-terminal M peptides, Mx and My. i represents an index for a percentage sequence identity range, and n is the number of corresponding polar heptad sites between peptides Mx and My that share sequence identity within range i and w is the weight associated with sequence identity range i.
The heptad scoring considers the sequence conservation or divergence at each of five polar heptad sites between a pair of M peptides. It begins by assessing the degree of residue-to-residue correspondence between the sequence patterns of two M peptides at a polar heptad site such that the order of residues in each sequence is preserved. The weights are simple scores and are based on the sequence identity range. For example, if there is <20% sequence identity at a heptad site between a pair of M peptides, the weight associated is −1 (penalty for low sequence identity), and for 20–29% sequence identity, the weight associated is +1 (heptad scoring key is in (Fig. 2). This assignment of a weight/score based on the sequence identity range at an individual heptad site score is repeated at all five polar sites, and the final pairwise heptad score between two M peptides is the sum of the scores at each heptad site; thus, the polar positions each equally contribute to the heptad score. The heptad scoring is heuristic and is thus not optimized but provides a satisfactory solution rapidly. It is a predictor of whether an Ab that recognizes sequence 1 will also recognize sequence 2.
The N-terminal sequences of M169 and M60 arranged in heptad repeats are shown alongside the helical wheel representation as parallel coiled-coil dimers in (Fig. 2. Also indicated is the general scheme of pairwise heptad scoring between M169 and M60. The sequence identity ranges associated with index i and their associated weights are given as the heptad scoring key in (Fig. 2. Higher sequence identity ranges were assigned higher weights. The range of the heptad scores was between -5 to 17.5. Higher scores indicate greater homology in the solvent-exposed heptad sites. A score of ≥10.5 was selected as a cutoff for distinguishing cross-reactive from non–cross-reactive peptide pairs.
Peptide Ags
The 50-aa peptide test Ags were custom synthesized by Peptide2Go (Manassas, VA) using solid-phase peptide synthesis, desalted, and supplied at 70% purity.
Recombinant NTC5 vaccine and synthetic peptide vaccines
Using the algorithms outlined above to identify cross-reactive peptide pairs, six M peptides (M4, M22, M165, M28, M88, and M78) were selected to include in a recombinant hybrid protein vaccine. Genes were synthesized (GenScript, Piscataway, NJ) to encode the six selected NTC5 M peptides in tandem. The coding sequence for the M4 peptide was included on the 5' and 3' regions based on previous studies, indicating that a duplicated peptide could function as a sacrificial peptide in the event that proteases preferentially degraded the N- or C-terminal regions (23). The synthetic gene, which also contained a T7 promoter and a 3' His tag, was ligated into pUC57, which was used to transform Escherichia coli carrying the T7 RNA pol. The recombinant protein was expressed and then purified by metal chelate chromatography, as previously reported in (23). Synthetic single peptide vaccines (M70, M121, M117, and M67) were formulated by conjugating each 50-aa peptide to keyhole limpet hemocyanin (KLH), using methods previously described in (16).
Immunizations
NTC5 vaccine
Three female New Zealand white rabbits (Charles River Laboratories, Wilmington, MA) were immunized i.m. with 200 µg of the NTC5 recombinant protein vaccine mixed with 25 µg LPS and then adsorbed to 280 µg aluminum hydroxide gel (Chemtrade, Berkeley Heights, NJ) at 0, 4, 8, and 17 wk. A booster injection of 200 µg with an equal amount of Addavax (InvivoGen, San Diego, CA) was given at 21 wk, and serum was obtained 2 wk after the final injection.
Single peptide vaccines
Groups of five mice each (male and female) were immunized i.m. with 30 µg of the indicated synthetic peptide–KLH conjugate adsorbed to 30 µg aluminum hydroxide gel at 0, 4, and 8 wk. The mice were a transgenic BALB/c strain maintained in our laboratory that expressed human C4BP and human factor H (28). Serum was obtained 3 wk after the final immunization and was pooled from each group of mice prior to performing ELISA with synthetic M peptides.
ELISAs
Rabbit and mouse antisera were assayed for Ab levels against M peptides by ELISA using previously described methods (29). ELISA titers were expressed as the inverse of the last dilution of antiserum that resulted in an OD of ≥0.15.
Bactericidal assays
Bactericidal assays were performed in quadruplicate using whole, nonimmune human blood, as previously described (30). Percentage of killing was calculated using the formula [(1 − (total CFU with test serum/total CFU with normal rabbit serum)) × 100]. Data were reported as the average percentage of killing in quadruplicate assays ±SD calculated after averaging the CFU in the four control samples.
Calculating potential vaccine coverage using epidemiologic data
Curated Strep A surveillance data were used to estimate the regional and potential disease coverage of M protein–based multivalent vaccines designed using the results of this study (31).
Institutional approvals
All animal procedures performed in this study were approved by the University of Tennessee Animal Care and Use Committee. The use of human blood in the bactericidal assays was approved by the Institutional Review Board (Ethics Committee) of the Free University of Brussels and the University of Tennessee Health Science Center.
Quantification and statistical analysis
The data were analyzed using nonparametric methods, Kruskal–Wallis for multiple group comparisons with Dunn–Bonferroni post hoc tests for pairwise comparisons with the IBM SPSS Statistics software (SPSS). All p values ≤0.05 were considered statistically significant.
The sensitivity, specificity, positive predictive value, and the Matthews correlation coefficient (MCC) were calculated to determine the effectiveness of the two-tiered approach and the revised approach in binary classification of cross-reactive and non–cross-reactive pairs. The MCC is a more useful and balanced metric of effectiveness of any binary classifier than F1 score or Accuracy when the dataset is imbalanced. Calculation of the MCC is based on all four elements of the confusion matrix: true positive, true negative, false positive, and false negative values. The MCC value has a range between −1 and 1, with −1, 0, and 1 indicating negative, random, and positive correlations, respectively, between observations and predictions.
Results
Predicting cross-reactivity among antigenic N-terminal M peptides of NTC5 using a two-tiered sequence- and structure-based method
Our vaccine design strategy focuses on the antigenic hypervariable N terminus of the coiled-coil M protein (Fig. 1). The heptad scoring scheme that forms the basis of cross-reactivity prediction between M peptides is shown in (Fig. 2 and described in detail in the Materials and Methods section. Based on prevalence data from a recent meta-analysis study (31), the 11 M types in the NTC5 cluster are epidemiologically important and account for 22, 18, 17, 14, 13, 10, and 7% of all circulating M types in Europe, North America, Asia, South America, Middle East, Africa, and Oceania, respectively (31) (Fig. 3A). The phylogenetic tree of the NTC5 comprising a total of 11 M types is shown in (Fig. 3B. The NTC5 50-mer N-terminal M peptides can differ by up to 75% in sequence identity, but all M types except M4 share a minimum of 55% sequence identity with at least one of the other 10 M types in the cluster. The N-terminal regions within the NTC5 peptides are predicted to have varying lengths of disordered structure followed by a repeating heptad motif indicative of a more structured coiled-coil (Supplemental Table I) (25).

Characteristics of N-terminal M peptides and clustering based on N-terminal 50-mer sequences. (A) Schematic representation of M protein coiled-coil dimer with the helical central rod region made up of increasingly variable repeat blocks (D, C, B, and A) from the C to the N terminus followed by the nonrepeat hypervariable N-terminal region. (B) Schematic representation of N-terminal hypervariable region of an M protein model generated in silico as a coiled-coil dimer displaying the interlocking of the hydrophobic residues of one monomer interface with a similar pattern on the other monomer. Hydrophobic residues are in blue and polar residues in magenta. (C) The 117 epidemiologically important GAS M types clustered based on the hypervariable N-terminal sequences (1–50 aa residues).
Characteristics of N-terminal M peptides and clustering based on N-terminal 50-mer sequences. (A) Schematic representation of M protein coiled-coil dimer with the helical central rod region made up of increasingly variable repeat blocks (D, C, B, and A) from the C to the N terminus followed by the nonrepeat hypervariable N-terminal region. (B) Schematic representation of N-terminal hypervariable region of an M protein model generated in silico as a coiled-coil dimer displaying the interlocking of the hydrophobic residues of one monomer interface with a similar pattern on the other monomer. Hydrophobic residues are in blue and polar residues in magenta. (C) The 117 epidemiologically important GAS M types clustered based on the hypervariable N-terminal sequences (1–50 aa residues).
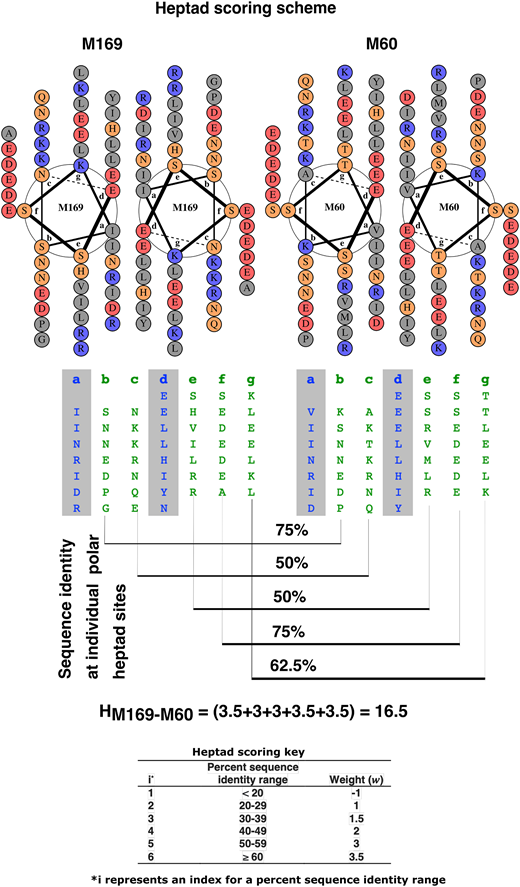
Heptad scoring scheme. Helical wheel representation of M169 and M60 (top) and heptad pattern of M169 and M60 (middle), along with heptad score (HM169–M60) between M169 and M60 and heptad scoring key (bottom). In the helical wheel representation, the nonpolar, polar, acidic, and basic residues are colored in gray, yellow, red, and blue, respectively. In the heptad pattern, the sites a and d (gray background) usually have hydrophobic residues and are shown in blue, and polar residues at the remaining sites are shown in green.
Heptad scoring scheme. Helical wheel representation of M169 and M60 (top) and heptad pattern of M169 and M60 (middle), along with heptad score (HM169–M60) between M169 and M60 and heptad scoring key (bottom). In the helical wheel representation, the nonpolar, polar, acidic, and basic residues are colored in gray, yellow, red, and blue, respectively. In the heptad pattern, the sites a and d (gray background) usually have hydrophobic residues and are shown in blue, and polar residues at the remaining sites are shown in green.

NTC5 epidemiology, M peptide phylogeny, and sequence patterns at each heptad site. (A) Estimated prevalence of the 11 NTC5 M types in different regions of the world. (B) Phylogenetic tree of the 50-mer NTC5 M peptides formed by neighbor joining and based on percentage sequence identity. (C) Multiple sequence alignment of the linear sequences of the NTC5 peptides at each heptad site using Clustal ω and visualized in Jalview. Hydrophobic, positively charged, negatively charged, polar, cysteine, glycine, proline, aromatic, and nonconserved residues are represented by blue, red, magenta, green, pink, orange, yellow, cyan, and white, respectively.
NTC5 epidemiology, M peptide phylogeny, and sequence patterns at each heptad site. (A) Estimated prevalence of the 11 NTC5 M types in different regions of the world. (B) Phylogenetic tree of the 50-mer NTC5 M peptides formed by neighbor joining and based on percentage sequence identity. (C) Multiple sequence alignment of the linear sequences of the NTC5 peptides at each heptad site using Clustal ω and visualized in Jalview. Hydrophobic, positively charged, negatively charged, polar, cysteine, glycine, proline, aromatic, and nonconserved residues are represented by blue, red, magenta, green, pink, orange, yellow, cyan, and white, respectively.
To identify a minimum number of vaccine peptides predicted to elicit Abs against all 11 NTC5 M types, we calculated the shared sequence identity and empirical pairwise heptad scores (17) (Materials and Methods) for each pair of NTC5 M peptides. The sequence arrangement at each heptad site for all NTC5 sequences is shown in (Fig. 3C, and the calculation of heptad scores between all possible nonredundant pairwise interactions of NTC5 M peptides that share at least ∼40% sequence identity is given in Table I. This analysis effectively identifies pairs of N-terminal M peptides with both moderate to high sequence identity and the desired feature of well-conserved polar amino acids located within the coiled-coil heptad domains. Based on these results, we selected six N-terminal 50-mer NTC5 peptides to include in a vaccine that were predicted to elicit cross-reactive Ab responses against all 11 NTC5 peptides. We hypothesized that this approach would be superior to using only linear sequence identity to predict peptides that will cross-react with heterologous M peptides in the same cluster.
Summary of 50 AA sequence identity, sequence identity between aligned coiled-coil domains, sequence identity between corresponding heptad sites, and heptad scores for pairs of NTC5 M peptides that share at least ∼40% sequence identity
| Pairwise Sequence Identity at Corresponding Heptad Sites between Mx and My | Number of Polar Heptad Sites Meeting Sequence Identity Range | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (Mx-My)a | 50-mer Sequence Identityb | N-Terminal Coiled-Coil Domain Sequence Identityc | A Hydrophobic | B Polar | C Polar | D Hydrophobic | E Polar | F Polar | G Polar | ≥20 and <30 (w = 1) | ≥30 and <40 (w = 1.5) | ≥40 and <50 (w = 2) | ≥50 and <60 (w = 3) | ≥60 (w = 3.5) | <20 (w = −1) | Heptad Score (H) |
| M8–M60 | 44.4 | 44.4 | 28.6 | 57.1 | 42.9 | 37.5 | 0.0 | 42.9 | 42.9 | 0 | 0 | 3 | 1 | 0 | 1 | 8.0 |
| M8–M78 | 61.8 | 63.8 | 28.6 | 42.9 | 28.6 | 28.6 | 28.6 | 50.0 | 11.1 | 2 | 0 | 1 | 1 | 0 | 1 | 6.0 |
| M8–M88d | 74.1 | 67.3 | 57.1 | 71.4 | 28.6 | 42.9 | 42.9 | 42.9 | 57.1 | 1 | 0 | 2 | 1 | 1 | 0 | 11.5 |
| M8–M165 | 47.1 | 44.7 | 57.1 | 42.9 | 8.3 | 57.1 | 42.9 | 66.7 | 33.3 | 0 | 1 | 2 | 0 | 1 | 1 | 8.0 |
| M8–M169d | 46.4 | 50.0 | 25.0 | 50.0 | 37.5 | 42.9 | 44.4 | 42.9 | 42.9 | 0 | 1 | 3 | 1 | 0 | 0 | 10.5 |
| M60–M22d | 51.9 | 52.0 | 20.0 | 42.9 | 42.9 | 50.0 | 22.2 | 71.4 | 57.1 | 1 | 0 | 2 | 1 | 1 | 0 | 11.5 |
| M60–M78 | 46.4 | 52.0 | 42.9 | 28.6 | 25.0 | 25.0 | 28.6 | 42.9 | 57.1 | 3 | 0 | 1 | 1 | 0 | 0 | 8.0 |
| M60–M88d | 52.0 | 46.0 | 57.1 | 71.4 | 28.6 | 25.0 | 42.9 | 42.9 | 57.1 | 1 | 0 | 2 | 1 | 1 | 0 | 11.5 |
| M60–M169d | 66.7 | 67.9 | 75.0 | 75.0 | 50.0 | 87.5 | 50.0 | 75.0 | 62.5 | 0 | 0 | 0 | 2 | 3 | 0 | 16.5 |
| M78–M88 | 51.8 | 54.2 | 42.9 | 42.9 | 14.3 | 50.0 | 50.0 | 14.3 | 42.9 | 0 | 0 | 2 | 1 | 0 | 2 | 5.0 |
| M78–M165 | 35.6 | 46.7 | 0.0 | 11.1 | 28.6 | 42.9 | 33.3 | 50.0 | 28.6 | 2 | 1 | 0 | 1 | 0 | 1 | 5.5 |
| M78–M169 | 62.3 | 57.1 | 42.9 | 28.6 | 28.6 | 28.6 | 28.6 | 42.9 | 57.1 | 3 | 0 | 1 | 1 | 0 | 0 | 8.0 |
| M88–M169d | 43.9 | 44.4 | 50.0 | 62.5 | 37.5 | 28.6 | 42.9 | 37.5 | 50.0 | 0 | 2 | 1 | 1 | 1 | 0 | 11.5 |
| M165–M176d | 57.1 | 62.2 | 66.7 | 42.9 | 71.4 | 57.1 | 100.0 | 33.3 | 66.7 | 0 | 1 | 1 | 0 | 3 | 0 | 14.0 |
| M22–M28 | 50.9 | 42.0 | 0.0 | 28.6 | 33.3 | 33.3 | 14.3 | 11.1 | 42.9 | 1 | 1 | 1 | 0 | 0 | 2 | 2.5 |
| M22–M109d | 61.8 | 63.3 | 42.9 | 71.4 | 57.1 | 57.1 | 42.9 | 57.1 | 57.1 | 0 | 0 | 1 | 3 | 1 | 0 | 14.5 |
| M28–M109 | 62.1 | 63.3 | 28.6 | 42.9 | 28.6 | 57.1 | 50 | 12.5 | 25 | 2 | 0 | 1 | 1 | 0 | 1 | 6.0 |
| Pairwise Sequence Identity at Corresponding Heptad Sites between Mx and My | Number of Polar Heptad Sites Meeting Sequence Identity Range | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (Mx-My)a | 50-mer Sequence Identityb | N-Terminal Coiled-Coil Domain Sequence Identityc | A Hydrophobic | B Polar | C Polar | D Hydrophobic | E Polar | F Polar | G Polar | ≥20 and <30 (w = 1) | ≥30 and <40 (w = 1.5) | ≥40 and <50 (w = 2) | ≥50 and <60 (w = 3) | ≥60 (w = 3.5) | <20 (w = −1) | Heptad Score (H) |
| M8–M60 | 44.4 | 44.4 | 28.6 | 57.1 | 42.9 | 37.5 | 0.0 | 42.9 | 42.9 | 0 | 0 | 3 | 1 | 0 | 1 | 8.0 |
| M8–M78 | 61.8 | 63.8 | 28.6 | 42.9 | 28.6 | 28.6 | 28.6 | 50.0 | 11.1 | 2 | 0 | 1 | 1 | 0 | 1 | 6.0 |
| M8–M88d | 74.1 | 67.3 | 57.1 | 71.4 | 28.6 | 42.9 | 42.9 | 42.9 | 57.1 | 1 | 0 | 2 | 1 | 1 | 0 | 11.5 |
| M8–M165 | 47.1 | 44.7 | 57.1 | 42.9 | 8.3 | 57.1 | 42.9 | 66.7 | 33.3 | 0 | 1 | 2 | 0 | 1 | 1 | 8.0 |
| M8–M169d | 46.4 | 50.0 | 25.0 | 50.0 | 37.5 | 42.9 | 44.4 | 42.9 | 42.9 | 0 | 1 | 3 | 1 | 0 | 0 | 10.5 |
| M60–M22d | 51.9 | 52.0 | 20.0 | 42.9 | 42.9 | 50.0 | 22.2 | 71.4 | 57.1 | 1 | 0 | 2 | 1 | 1 | 0 | 11.5 |
| M60–M78 | 46.4 | 52.0 | 42.9 | 28.6 | 25.0 | 25.0 | 28.6 | 42.9 | 57.1 | 3 | 0 | 1 | 1 | 0 | 0 | 8.0 |
| M60–M88d | 52.0 | 46.0 | 57.1 | 71.4 | 28.6 | 25.0 | 42.9 | 42.9 | 57.1 | 1 | 0 | 2 | 1 | 1 | 0 | 11.5 |
| M60–M169d | 66.7 | 67.9 | 75.0 | 75.0 | 50.0 | 87.5 | 50.0 | 75.0 | 62.5 | 0 | 0 | 0 | 2 | 3 | 0 | 16.5 |
| M78–M88 | 51.8 | 54.2 | 42.9 | 42.9 | 14.3 | 50.0 | 50.0 | 14.3 | 42.9 | 0 | 0 | 2 | 1 | 0 | 2 | 5.0 |
| M78–M165 | 35.6 | 46.7 | 0.0 | 11.1 | 28.6 | 42.9 | 33.3 | 50.0 | 28.6 | 2 | 1 | 0 | 1 | 0 | 1 | 5.5 |
| M78–M169 | 62.3 | 57.1 | 42.9 | 28.6 | 28.6 | 28.6 | 28.6 | 42.9 | 57.1 | 3 | 0 | 1 | 1 | 0 | 0 | 8.0 |
| M88–M169d | 43.9 | 44.4 | 50.0 | 62.5 | 37.5 | 28.6 | 42.9 | 37.5 | 50.0 | 0 | 2 | 1 | 1 | 1 | 0 | 11.5 |
| M165–M176d | 57.1 | 62.2 | 66.7 | 42.9 | 71.4 | 57.1 | 100.0 | 33.3 | 66.7 | 0 | 1 | 1 | 0 | 3 | 0 | 14.0 |
| M22–M28 | 50.9 | 42.0 | 0.0 | 28.6 | 33.3 | 33.3 | 14.3 | 11.1 | 42.9 | 1 | 1 | 1 | 0 | 0 | 2 | 2.5 |
| M22–M109d | 61.8 | 63.3 | 42.9 | 71.4 | 57.1 | 57.1 | 42.9 | 57.1 | 57.1 | 0 | 0 | 1 | 3 | 1 | 0 | 14.5 |
| M28–M109 | 62.1 | 63.3 | 28.6 | 42.9 | 28.6 | 57.1 | 50 | 12.5 | 25 | 2 | 0 | 1 | 1 | 0 | 1 | 6.0 |
Nonredundant M type pairs (Mx-My) that share ∼40% sequence identity between their 50-mer N-terminal regions or between their N-terminal coiled-coil domains.
Sequence identity between the 50-mer N-terminal regions of Mx and My.
Sequence identity between the N-terminal coiled-coil domains of 50-mer Mx and My peptides.
Pairs that are predicted to cross-react.
Heptad scores of the pairs predicted to cross-react are in bold. w indicates the weight associated with the sequence identity range.
Recombinant NTC5 vaccine elicits cross-reactive Abs against predicted nonvaccine M types
A recombinant vaccine was constructed that contained six N-terminal 50 aa peptides from NTC5 predicted to cross-react with the five nonvaccine M types in the NTC5 cluster (Fig. 4A). Four of the six vaccine peptides (M22, M28, M88, and M165) share sequence identity of ≥40% and a pairwise heptad score ≥10.5 with one or more heterologous M peptides in the cluster. Two of the peptides (M4 and M78) were considered “type-specific” because the analyses indicated that they did not achieve the threshold for sequence identity or heptad score. The immune sera from three rabbits immunized with the NTC5 vaccine contained high titers of Abs against all 11 NTC5 M peptides (Fig. 4B). To determine the functional activity of the M protein Abs evoked by the six-peptide NTC5 vaccine, in vitro opsonophagocytic killing assays were performed with each of the six vaccine M types, three of the five nonvaccine M types in the NTC5 cluster (M176 was not available for these assays, and M169 consistently failed to achieve sufficient growth in blood samples from multiple donors), and four non-NTC5 nonvaccine M types. The NTC5 vaccine antisera promoted opsonization and killing of all eight NTC5 M types that were tested in the in vitro whole blood killing assays (Table II). No significant bactericidal activity was observed against the four non-NTC5 nonvaccine M types.

NTC5 vaccine construct with the observed cross-reactive immune responses in rabbits immunized with the multivalent vaccine. (A) Recombinant NTC5 M peptide–based vaccine with expected coverage of nonvaccine M types based on sequence identity of >40% and heptad score threshold ≥10.5. (B) Rabbit serum ELISA titers against NTC5 50 aa vaccine and nonvaccine M peptides and non-NTC5 50 aa M peptides. Data analysis performed using Kruskal–Wallis test with Dunn–Bonferroni correction; ***p ≤ 0.001. N.S., not statistically significant, with p > 0.05 indicating no difference in means.
NTC5 vaccine construct with the observed cross-reactive immune responses in rabbits immunized with the multivalent vaccine. (A) Recombinant NTC5 M peptide–based vaccine with expected coverage of nonvaccine M types based on sequence identity of >40% and heptad score threshold ≥10.5. (B) Rabbit serum ELISA titers against NTC5 50 aa vaccine and nonvaccine M peptides and non-NTC5 50 aa M peptides. Data analysis performed using Kruskal–Wallis test with Dunn–Bonferroni correction; ***p ≤ 0.001. N.S., not statistically significant, with p > 0.05 indicating no difference in means.
Serum bactericidal Abs evoked in rabbits by the NTC5 vaccine, as determined in assays whole human blood
| Percentage Killing (±SD) Promoted by NTC5 Vaccine Rabbit Serum | |||
|---|---|---|---|
| M Type | Rabbit A | Rabbit B | Rabbit C |
| Vaccine | |||
| M4 | 43 ± 34 | 64 ± 13 | 59 ± 16 |
| M22 | 73 ± 33 | 63 ± 20 | 94 ± 9 |
| M165 | 53 ± 1 | 30 ± 27 | 7 ± 4 |
| M28 | 36 ± 19 | 63 ± 3 | 39 ± 20 |
| M88 | 50 ± 22 | 52 ± 20 | 59 ± 21 |
| M78 | 65 ± 35 | 60 ± 16 | 65 ± 21 |
| Nonvaccine | |||
| M109 | 41 ± 12 | 45 ± 4 | 48 ± 10 |
| M60 | 0 | 37 ± 19 | 42 ± 17 |
| M8 | 43 ± 21 | 50 ± 21 | 35 ± 18 |
| M169 | N.D. | N.D. | N.D. |
| M176 | N.D. | N.D. | N.D. |
| Non-NTC5 | |||
| M12 | 0 | 0 | 0 |
| M77 | 0 | 0 | 5 ± 23 |
| M1 | 0 | 0 | 0 |
| M2 | 13 ± 24 | 0 | 8 ± 24 |
| Percentage Killing (±SD) Promoted by NTC5 Vaccine Rabbit Serum | |||
|---|---|---|---|
| M Type | Rabbit A | Rabbit B | Rabbit C |
| Vaccine | |||
| M4 | 43 ± 34 | 64 ± 13 | 59 ± 16 |
| M22 | 73 ± 33 | 63 ± 20 | 94 ± 9 |
| M165 | 53 ± 1 | 30 ± 27 | 7 ± 4 |
| M28 | 36 ± 19 | 63 ± 3 | 39 ± 20 |
| M88 | 50 ± 22 | 52 ± 20 | 59 ± 21 |
| M78 | 65 ± 35 | 60 ± 16 | 65 ± 21 |
| Nonvaccine | |||
| M109 | 41 ± 12 | 45 ± 4 | 48 ± 10 |
| M60 | 0 | 37 ± 19 | 42 ± 17 |
| M8 | 43 ± 21 | 50 ± 21 | 35 ± 18 |
| M169 | N.D. | N.D. | N.D. |
| M176 | N.D. | N.D. | N.D. |
| Non-NTC5 | |||
| M12 | 0 | 0 | 0 |
| M77 | 0 | 0 | 5 ± 23 |
| M1 | 0 | 0 | 0 |
| M2 | 13 ± 24 | 0 | 8 ± 24 |
M169 consistently failed to achieve sufficient growth in blood samples from multiple donors, and 176 was not available for these assays; thus, percentage killing in the presence of vaccine Abs could not be assessed for these M types. Rabbit designations (A, B, and C) correspond to the order of the three bars in (Fig 4.
N.D., not determined.
Cross-reactive Ab binding among heterologous M peptides also depends on the coiled-coil propensity and coiled-coil length
We next examined the utility of the two-tiered criterion (pairwise sequence identity ≥40% and heptad score ≥10.5 beyond the N-terminal M peptides in NTC5 (this work) and NTC6 (17), which mostly had long ordered coiled-coil domains. We wanted to test if the two-tiered approach would also be suitable to predict cross-reactivity of M peptides that had shorter and more disordered coiled-coil domains. We constructed individual peptide vaccines from the N-terminal 50-mers of M70 (NTC7), M121 (NTC7), M117 (NTC1), and M67 (NTC4), each covalently linked to KLH as a carrier. These M peptides were selected based on their shared sequence identity with many other M peptides and aspects of their predicted three-dimensional structures: M117 and M67 are predicted to have relatively long and ordered coiled-coil domains, whereas M70 and M121 are predicted to have short and low probability coiled-coil domains (Table III). Groups of five mice each were immunized with a single peptide vaccine (Fig. 5A), and serum from each group was pooled. ELISA was performed using 32 different N-terminal 50-mer M peptides that share significant sequence identity (>40%) with at least one of the four vaccine peptides and nine additional M peptides that share low sequence identity (i.e., ≤40%) with all of the vaccine peptides (Table III).

Immune responses of mice to single M peptide vaccines, which served as the basis for refined criteria to select cross-reactive vaccine peptides. (A) Four groups of five BALB/c mice each immunized with single 50 aa vaccine peptides M70, M121, M117, and M67. Peptides M70 and M121 have 6 and 22% coiled-coil probability, respectively, and the respective lengths of their predicted coiled-coil domains are 23 and 25 aa long and are thus both represented as predominantly disordered peptides with short coiled-coil domains. Peptides M117 and M67 have 67 and 68.5% coiled-coil probability, respectively, and the respective lengths of their predicted coiled-coil domains are 41 and 40 aa long and are thus both represented as long structured coiled-coil peptides. (B) Immune responses in mice as determined by ELISA against 41 M peptides partitioned into the following groups: 5, 14, 6, and 7 M peptides that share >40% sequence identity with M70 (M70 group), M121 (M121 group), M117 (M117 group), and M67 (M67 group), respectively, and immune responses as determined by ELISA against 9 M peptides that share <40% sequence identity all four vaccine peptides (low sequence identity group). M70, M121, M117, and M67 Ab titers are represented by black, red, blue, and green bars. (C) Vaccine and nonvaccine peptide pairs divided into four classes based on predicted coiled-coil length and coiled-coil propensity. (D) Ab binding (ELISA OD) versus shared 50-mer sequence identity, (E) Ab binding (ELISA OD) versus shared coiled-coil domain sequence identity, and (F) Ab binding (ELISA OD) versus pairwise heptad score.
Immune responses of mice to single M peptide vaccines, which served as the basis for refined criteria to select cross-reactive vaccine peptides. (A) Four groups of five BALB/c mice each immunized with single 50 aa vaccine peptides M70, M121, M117, and M67. Peptides M70 and M121 have 6 and 22% coiled-coil probability, respectively, and the respective lengths of their predicted coiled-coil domains are 23 and 25 aa long and are thus both represented as predominantly disordered peptides with short coiled-coil domains. Peptides M117 and M67 have 67 and 68.5% coiled-coil probability, respectively, and the respective lengths of their predicted coiled-coil domains are 41 and 40 aa long and are thus both represented as long structured coiled-coil peptides. (B) Immune responses in mice as determined by ELISA against 41 M peptides partitioned into the following groups: 5, 14, 6, and 7 M peptides that share >40% sequence identity with M70 (M70 group), M121 (M121 group), M117 (M117 group), and M67 (M67 group), respectively, and immune responses as determined by ELISA against 9 M peptides that share <40% sequence identity all four vaccine peptides (low sequence identity group). M70, M121, M117, and M67 Ab titers are represented by black, red, blue, and green bars. (C) Vaccine and nonvaccine peptide pairs divided into four classes based on predicted coiled-coil length and coiled-coil propensity. (D) Ab binding (ELISA OD) versus shared 50-mer sequence identity, (E) Ab binding (ELISA OD) versus shared coiled-coil domain sequence identity, and (F) Ab binding (ELISA OD) versus pairwise heptad score.
Pairwise analysis of total sequence identity, coiled-coil sequence identity, and heptad scores of the 32 peptides that share significant sequence identity with at least one of the monovalent vaccine peptides (M70, M121, M117, and M67), and additionally, nine peptides that share low sequence identity with all of the monovalent vaccine peptides
| M Types | Coiled-Coil Length (No. of Residues) | Average Coiled- Coil Probability | 50-mer Sequence Identity with | Sequence Identity between Coiled–Coil Domains | Pairwise Heptad Score | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M70 | M121 | M117 | M67 | M70 | M121 | M117 | M67 | M70 | M121 | M117 | M67 | |||
| M70a | 23 | 6.0 | 100.0 | 19.3 | 24.5 | 2.2 | 100.0 | 15.4 | 20.9 | 15.0 | 17.5 | 5.5 | −2.5 | −2.5 |
| M33 | 50 | 15.4 | 68.6 | 21.5 | 20.8 | 12.3 | 36.0 | 7.3 | 18.9 | 16.1 | 6.0 | −3.0 | −3.0 | −1.0 |
| M225 | 19 | 73.8 | 56.9 | 13.3 | 24.5 | 15.1 | 34.0 | 18.0 | 22.6 | 17.7 | 5.0 | −3.0 | 1.0 | 4.5 |
| M230 | 50 | 73.1 | 54.9 | 20.3 | 18.3 | 12.3 | 32.0 | 20.0 | 20.0 | 12.9 | 4.0 | −3.0 | −3.0 | −0.5 |
| M108 | 28 | 14.7 | 80.0 | 19.3 | 22.6 | 17.5 | 64.3 | 16.1 | 16.3 | 20.9 | 12.5 | 5.0 | −5.0 | 0.0 |
| M121a | 25 | 22.2 | 19.3 | 100.0 | 27.1 | 19.2 | 15.4 | 100.0 | 22.0 | 22.7 | 5.5 | 17.5 | −3.0 | −2.5 |
| M52 | 40 | 25.2 | 23.6 | 62.7 | 30.0 | 17.5 | 12.5 | 39.0 | 32.0 | 16.3 | −2.0 | 6.5 | −0.5 | −5.0 |
| M64 | 40 | 40.3 | 26.7 | 58.8 | 17.5 | 21.7 | 24.4 | 45.0 | 18.9 | 26.0 | −3.0 | 8.5 | 1.5 | 1.0 |
| M43 | 27 | 43.7 | 22.6 | 38.5 | 23.0 | 22.4 | 17.1 | 44.4 | 14.6 | 17.1 | 3.0 | 3.5 | −2.0 | −2.5 |
| M72 | 33 | 49.8 | 23.3 | 34.5 | 24.6 | 23.6 | 15.2 | 42.4 | 18.2 | 25.0 | 2.0 | 8.0 | −0.5 | −0.5 |
| M98 | 46 | 49.9 | 19.6 | 47.3 | 30.0 | 23.3 | 15.2 | 34.8 | 27.5 | 19.6 | −5.0 | 7.0 | −1.0 | −0.5 |
| M80 | 27 | 47.4 | 22.6 | 40.7 | 24.6 | 25.9 | 26.7 | 48.3 | 20.9 | 30.0 | 5.0 | 10.0 | −1.0 | −2.5 |
| M123 | 25 | 29.1 | 19.4 | 44.2 | 24.6 | 14.7 | 14.3 | 51.9 | 26.8 | 25.0 | 1.0 | 10.0 | −3.0 | 0.0 |
| M192 | 45 | 11.4 | 18.5 | 62.7 | 26.2 | 16.2 | 14.8 | 60.7 | 29.5 | 18.6 | 1.0 | 14.0 | −0.5 | −5.0 |
| M101 | 27 | 21.1 | 16.4 | 63.5 | 22.0 | 12.9 | 14.3 | 85.2 | 19.5 | 19.6 | 5.0 | 17.0 | −5.0 | 0.0 |
| M119 | 34 | 13.1 | 20.0 | 66.0 | 22.7 | 13.9 | 21.1 | 47.1 | 29.5 | 16.4 | 5.0 | 10.5 | −5.0 | −3.0 |
| M83 | 31 | 42.4 | 21.3 | 54.7 | 27.1 | 16.2 | 21.2 | 58.1 | 28.9 | 16.3 | −1.0 | 11.5 | 2.0 | −2.5 |
| M186 | 27 | 46.3 | 15.9 | 51.0 | 22.4 | 21.1 | 15.8 | 51.7 | 9.8 | 17.5 | 3.0 | 14.5 | −5.0 | 3.0 |
| M178 | 44 | 46.4 | 15.9 | 41.5 | 12.8 | 20.3 | 13.9 | 50.0 | 21.4 | 17.0 | 1.0 | 13.5 | −2.0 | 0.0 |
| M117a | 41 | 66.7 | 24.5 | 27.1 | 100.0 | 29.2 | 20.9 | 22.0 | 100.0 | 25.0 | −2.5 | −3.0 | 17.5 | −0.5 |
| M158 | 45 | 63.7 | 16.4 | 21.3 | 44.6 | 34.6 | 17.9 | 23.8 | 53.7 | 39.0 | 3.0 | −2.5 | 12.0 | 10.0 |
| M92 | 40 | 62.6 | 18.9 | 22.8 | 53.8 | 32.7 | 19.5 | 9.8 | 52.4 | 31.7 | −2.5 | −3.0 | 7.5 | 3.0 |
| M113 | 41 | 69.7 | 20.6 | 23.8 | 68.0 | 39.2 | 17.1 | 20.0 | 65.9 | 35.7 | −5.0 | −5.0 | 15.0 | 4.0 |
| M27 | 43 | 67.0 | 11.3 | 22.4 | 47.1 | 31.5 | 14.3 | 16.3 | 44.2 | 27.7 | −5.0 | −2.5 | 9.0 | 3.5 |
| M76 | 43 | 62.3 | 11.1 | 20.7 | 48.1 | 37.7 | 14.0 | 20.9 | 45.5 | 34.8 | 0.0 | 2.5 | 11.5 | 4.0 |
| M67a | 40 | 68.5 | 2.2 | 19.2 | 29.2 | 100.0 | 15.0 | 22.7 | 25.0 | 100.0 | −2.5 | −2.5 | −0.5 | 17.5 |
| M11 | 36 | 36.2 | 17.4 | 17.2 | 26.5 | 40.0 | 17.9 | 15.0 | 25.5 | 51.2 | −1.0 | 3.5 | 4.0 | 6.0 |
| M85 | 38 | 42.8 | 8.3 | 7.9 | 33.3 | 43.4 | 17.1 | 17.1 | 34.1 | 55.0 | −1.0 | −3.0 | 3.5 | 10.0 |
| M42 | 50 | 85.4 | 14.3 | 20.0 | 25.9 | 46.3 | 11.3 | 17.2 | 28.0 | 40.7 | −5.0 | 1.0 | 5.0 | 5.0 |
| M65 | 40 | 65.6 | 12.9 | 24.1 | 37.0 | 80.4 | 12.5 | 15.6 | 34.1 | 78.0 | −0.5 | −5.0 | 6.0 | 17.5 |
| M44 | 44 | 67.9 | 15.6 | 22.4 | 32.3 | 66.0 | 18.2 | 25.0 | 29.3 | 63.6 | −5.0 | −3.0 | 1.5 | 14.5 |
| M13 | 35 | 26.6 | 17.9 | 23.3 | 60.4 | 41.5 | 16.3 | 23.1 | 53.7 | 40.0 | 2.5 | 3.0 | 13.0 | 8.5 |
| M99 | 47 | 83.4 | 6.4 | 17.3 | 32.1 | 27.6 | 5.9 | 20.0 | 30.8 | 31.2 | −5.0 | −3.0 | 2.5 | 3.0 |
| M106 | 46 | 64.9 | 10.0 | 20.4 | 39.6 | 34.0 | 14.8 | 17.4 | 36.7 | 32.7 | −5.0 | −2.5 | 1.5 | 4.5 |
| M91 | 24 | 38.6 | 4.5 | 43.1 | 22.6 | 16.7 | 12.9 | 38.5 | 19.5 | 15.6 | 1.0 | 9.5 | 0.5 | −2.5 |
| M53 | 30 | 47.9 | 22.0 | 30.9 | 28.1 | 29.1 | 13.9 | 33.3 | 20.5 | 27.5 | 1.0 | 1.0 | −2.0 | −2.5 |
| M56 | 37 | 6.2 | 19.6 | 44.6 | 23.3 | 4.8 | 1.8 | 33.3 | 25.0 | 11.1 | 0.0 | 3.0 | 4.5 | −2.5 |
| M120 | 35 | 3.1 | 6.0 | 35.7 | 22.7 | 12.9 | 1.8 | 32.4 | 26.0 | 14.3 | −1.0 | 4.0 | 3.0 | −2.5 |
| M224 | 48 | 70.7 | 23.2 | 16.2 | 20.6 | 22.4 | 9.6 | 10.9 | 16.4 | 17.5 | −5.0 | −5.0 | −3.0 | 1.0 |
| M191 | 49 | 43.7 | 4.5 | 24.1 | 37.7 | 15.1 | 16.0 | 17.0 | 30.8 | 14.9 | −3.0 | −1.0 | 5.5 | 0.0 |
| M182 | 29 | 67.6 | 11.6 | 25.4 | 42.3 | 26.6 | 11.4 | 22.7 | 40.0 | 24.1 | −2.5 | −0.5 | 5.0 | 6.0 |
| M Types | Coiled-Coil Length (No. of Residues) | Average Coiled- Coil Probability | 50-mer Sequence Identity with | Sequence Identity between Coiled–Coil Domains | Pairwise Heptad Score | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M70 | M121 | M117 | M67 | M70 | M121 | M117 | M67 | M70 | M121 | M117 | M67 | |||
| M70a | 23 | 6.0 | 100.0 | 19.3 | 24.5 | 2.2 | 100.0 | 15.4 | 20.9 | 15.0 | 17.5 | 5.5 | −2.5 | −2.5 |
| M33 | 50 | 15.4 | 68.6 | 21.5 | 20.8 | 12.3 | 36.0 | 7.3 | 18.9 | 16.1 | 6.0 | −3.0 | −3.0 | −1.0 |
| M225 | 19 | 73.8 | 56.9 | 13.3 | 24.5 | 15.1 | 34.0 | 18.0 | 22.6 | 17.7 | 5.0 | −3.0 | 1.0 | 4.5 |
| M230 | 50 | 73.1 | 54.9 | 20.3 | 18.3 | 12.3 | 32.0 | 20.0 | 20.0 | 12.9 | 4.0 | −3.0 | −3.0 | −0.5 |
| M108 | 28 | 14.7 | 80.0 | 19.3 | 22.6 | 17.5 | 64.3 | 16.1 | 16.3 | 20.9 | 12.5 | 5.0 | −5.0 | 0.0 |
| M121a | 25 | 22.2 | 19.3 | 100.0 | 27.1 | 19.2 | 15.4 | 100.0 | 22.0 | 22.7 | 5.5 | 17.5 | −3.0 | −2.5 |
| M52 | 40 | 25.2 | 23.6 | 62.7 | 30.0 | 17.5 | 12.5 | 39.0 | 32.0 | 16.3 | −2.0 | 6.5 | −0.5 | −5.0 |
| M64 | 40 | 40.3 | 26.7 | 58.8 | 17.5 | 21.7 | 24.4 | 45.0 | 18.9 | 26.0 | −3.0 | 8.5 | 1.5 | 1.0 |
| M43 | 27 | 43.7 | 22.6 | 38.5 | 23.0 | 22.4 | 17.1 | 44.4 | 14.6 | 17.1 | 3.0 | 3.5 | −2.0 | −2.5 |
| M72 | 33 | 49.8 | 23.3 | 34.5 | 24.6 | 23.6 | 15.2 | 42.4 | 18.2 | 25.0 | 2.0 | 8.0 | −0.5 | −0.5 |
| M98 | 46 | 49.9 | 19.6 | 47.3 | 30.0 | 23.3 | 15.2 | 34.8 | 27.5 | 19.6 | −5.0 | 7.0 | −1.0 | −0.5 |
| M80 | 27 | 47.4 | 22.6 | 40.7 | 24.6 | 25.9 | 26.7 | 48.3 | 20.9 | 30.0 | 5.0 | 10.0 | −1.0 | −2.5 |
| M123 | 25 | 29.1 | 19.4 | 44.2 | 24.6 | 14.7 | 14.3 | 51.9 | 26.8 | 25.0 | 1.0 | 10.0 | −3.0 | 0.0 |
| M192 | 45 | 11.4 | 18.5 | 62.7 | 26.2 | 16.2 | 14.8 | 60.7 | 29.5 | 18.6 | 1.0 | 14.0 | −0.5 | −5.0 |
| M101 | 27 | 21.1 | 16.4 | 63.5 | 22.0 | 12.9 | 14.3 | 85.2 | 19.5 | 19.6 | 5.0 | 17.0 | −5.0 | 0.0 |
| M119 | 34 | 13.1 | 20.0 | 66.0 | 22.7 | 13.9 | 21.1 | 47.1 | 29.5 | 16.4 | 5.0 | 10.5 | −5.0 | −3.0 |
| M83 | 31 | 42.4 | 21.3 | 54.7 | 27.1 | 16.2 | 21.2 | 58.1 | 28.9 | 16.3 | −1.0 | 11.5 | 2.0 | −2.5 |
| M186 | 27 | 46.3 | 15.9 | 51.0 | 22.4 | 21.1 | 15.8 | 51.7 | 9.8 | 17.5 | 3.0 | 14.5 | −5.0 | 3.0 |
| M178 | 44 | 46.4 | 15.9 | 41.5 | 12.8 | 20.3 | 13.9 | 50.0 | 21.4 | 17.0 | 1.0 | 13.5 | −2.0 | 0.0 |
| M117a | 41 | 66.7 | 24.5 | 27.1 | 100.0 | 29.2 | 20.9 | 22.0 | 100.0 | 25.0 | −2.5 | −3.0 | 17.5 | −0.5 |
| M158 | 45 | 63.7 | 16.4 | 21.3 | 44.6 | 34.6 | 17.9 | 23.8 | 53.7 | 39.0 | 3.0 | −2.5 | 12.0 | 10.0 |
| M92 | 40 | 62.6 | 18.9 | 22.8 | 53.8 | 32.7 | 19.5 | 9.8 | 52.4 | 31.7 | −2.5 | −3.0 | 7.5 | 3.0 |
| M113 | 41 | 69.7 | 20.6 | 23.8 | 68.0 | 39.2 | 17.1 | 20.0 | 65.9 | 35.7 | −5.0 | −5.0 | 15.0 | 4.0 |
| M27 | 43 | 67.0 | 11.3 | 22.4 | 47.1 | 31.5 | 14.3 | 16.3 | 44.2 | 27.7 | −5.0 | −2.5 | 9.0 | 3.5 |
| M76 | 43 | 62.3 | 11.1 | 20.7 | 48.1 | 37.7 | 14.0 | 20.9 | 45.5 | 34.8 | 0.0 | 2.5 | 11.5 | 4.0 |
| M67a | 40 | 68.5 | 2.2 | 19.2 | 29.2 | 100.0 | 15.0 | 22.7 | 25.0 | 100.0 | −2.5 | −2.5 | −0.5 | 17.5 |
| M11 | 36 | 36.2 | 17.4 | 17.2 | 26.5 | 40.0 | 17.9 | 15.0 | 25.5 | 51.2 | −1.0 | 3.5 | 4.0 | 6.0 |
| M85 | 38 | 42.8 | 8.3 | 7.9 | 33.3 | 43.4 | 17.1 | 17.1 | 34.1 | 55.0 | −1.0 | −3.0 | 3.5 | 10.0 |
| M42 | 50 | 85.4 | 14.3 | 20.0 | 25.9 | 46.3 | 11.3 | 17.2 | 28.0 | 40.7 | −5.0 | 1.0 | 5.0 | 5.0 |
| M65 | 40 | 65.6 | 12.9 | 24.1 | 37.0 | 80.4 | 12.5 | 15.6 | 34.1 | 78.0 | −0.5 | −5.0 | 6.0 | 17.5 |
| M44 | 44 | 67.9 | 15.6 | 22.4 | 32.3 | 66.0 | 18.2 | 25.0 | 29.3 | 63.6 | −5.0 | −3.0 | 1.5 | 14.5 |
| M13 | 35 | 26.6 | 17.9 | 23.3 | 60.4 | 41.5 | 16.3 | 23.1 | 53.7 | 40.0 | 2.5 | 3.0 | 13.0 | 8.5 |
| M99 | 47 | 83.4 | 6.4 | 17.3 | 32.1 | 27.6 | 5.9 | 20.0 | 30.8 | 31.2 | −5.0 | −3.0 | 2.5 | 3.0 |
| M106 | 46 | 64.9 | 10.0 | 20.4 | 39.6 | 34.0 | 14.8 | 17.4 | 36.7 | 32.7 | −5.0 | −2.5 | 1.5 | 4.5 |
| M91 | 24 | 38.6 | 4.5 | 43.1 | 22.6 | 16.7 | 12.9 | 38.5 | 19.5 | 15.6 | 1.0 | 9.5 | 0.5 | −2.5 |
| M53 | 30 | 47.9 | 22.0 | 30.9 | 28.1 | 29.1 | 13.9 | 33.3 | 20.5 | 27.5 | 1.0 | 1.0 | −2.0 | −2.5 |
| M56 | 37 | 6.2 | 19.6 | 44.6 | 23.3 | 4.8 | 1.8 | 33.3 | 25.0 | 11.1 | 0.0 | 3.0 | 4.5 | −2.5 |
| M120 | 35 | 3.1 | 6.0 | 35.7 | 22.7 | 12.9 | 1.8 | 32.4 | 26.0 | 14.3 | −1.0 | 4.0 | 3.0 | −2.5 |
| M224 | 48 | 70.7 | 23.2 | 16.2 | 20.6 | 22.4 | 9.6 | 10.9 | 16.4 | 17.5 | −5.0 | −5.0 | −3.0 | 1.0 |
| M191 | 49 | 43.7 | 4.5 | 24.1 | 37.7 | 15.1 | 16.0 | 17.0 | 30.8 | 14.9 | −3.0 | −1.0 | 5.5 | 0.0 |
| M182 | 29 | 67.6 | 11.6 | 25.4 | 42.3 | 26.6 | 11.4 | 22.7 | 40.0 | 24.1 | −2.5 | −0.5 | 5.0 | 6.0 |
Vaccine peptides.
(Fig. 5B shows the observed levels of cross-reactivity of the vaccine antisera with the nonvaccine peptides that share considerable sequence identity with the vaccine peptides M70, M121, M117, and M67 and with the nine nonvaccine peptides that do not share significant sequence identity with any of the vaccine peptides, respectively. We obtained a MCC of 0.42 for the two-tiered approach, indicating a positive correlation between the cross-reactivity prediction and the observed Ab cross-reactivity (ELISA OD ≥ 0.3) (Supplemental Table II). Intermediate (OD: 0.3–0.5) to higher (OD > 0.5) levels of cross-reactive Ab binding were observed for vaccine–nonvaccine M peptide pairs that share sequence identity and heptad scores above the threshold of 40% and 10.5, respectively. None of the four vaccine antisera cross-reacted with the nine nonvaccine peptides that share fewer than 40% sequence identity with the vaccine peptides. Unexpectedly, the M121 vaccine peptide, which shares considerable sequence identity (41–66%) with 13/41 of the heterologous M peptides, did not elicit cross-reactive Abs, including Abs against 5/13 M peptides with which it also shares heptad scores above 10.5.
To improve the cross-reactivity prediction algorithm and to understand the source of discrepancy between observed and predicted M121 vaccine cross-reactivity, we next divided the assays into four classes based on the length of the predicted coiled-coil domains and average coiled-coil probabilities of the peptides (Fig. 5C, Supplemental Table II) as follows.
Class 1
One or both peptides in the pair involved in the ELISA (i.e., either the peptide coating the plate or the vaccine peptide for which the primary Ab is specific) are predicted to have a short coiled-coil domain (<35 aa) and low average coiled-coil probability (<50%).
Class 2
Both peptides in the pair have an average predicted coiled-coil probability >50%, but at least one is predicted to have a short coiled-coil domain (<35 aa).
Class 3
Both peptides involved in the pair are predicted to have long coiled-coil domains (>35 aa), but at least one is predicted to have a low coiled-coil probability (<50%).
Class 4
Both peptides are predicted to have long coiled-coil domains (>35 aa) with high average coiled-coil probabilities (>50%).
For class 4, in which both peptides in the pair are predicted to be long, stable coiled-coils, the observed pair cross-reactivity correlated significantly with the three parameters, namely pairwise sequence identity between aligned 50-mers, pairwise sequence identity between the aligned coiled-coil domains, and pairwise heptad scores (Fig. 5D–F). However, for classes 1 and 3, in which there was a possibility of a short coiled-coil domain or low average coiled-coil probability or both, there was no significant correlation between pair cross-reactivity and these parameters. Also, for any given sequence identity, higher cross-reactivity was found for class 4 peptides than classes 1 and 3, suggesting a stronger and a more robust cross-reactive immune response when both peptides in the pair contain longer structured coiled-coils. Class 2 had insufficient data points to generate meaningful correlation coefficients.
Overall, we found that despite considerable shared sequence identity within overlapping 50-mers (for example, M70–M33, M70–M225, and M121-M64), if one peptide in the pair is predicted to have a short coiled-coil domain and/or very low coiled-coil probability, then these peptides do not cross-react. This implies that, even in cases of substantial sequence identity, peptide conformation is an important predictor of cross-reactive Ab binding. The predicted coiled-coil domain lengths and average coiled-coil probabilities, when taken together with the sequence identity and heptad scores, permits a realistic and rapid assessment of the degree of structural similarity between two N-terminal M peptides. We deduced that the false positives for the two-tiered approach resulted from mismatches in the coiled-coil length or average coiled-coil propensity between the pair of M peptides. The false negatives may have resulted from incorrect assignment of the heptad register that led to falsely low heptad scores, which is plausible for M peptides that have lower levels of coiled-coil probability.
Revised criteria of M type cross-reactivity prediction have very high precision and recall
Based on these findings, we propose a more stringent set of structural criteria designed to predict with higher specificity the likelihood of eliciting significant levels of cross-reactive Abs between a pair of heterologous M peptides. The revised rules are summarized in Table IV. The revised criteria consider the length of the predicted coiled-coil region and the degree of coiled-coil propensity plus the sequence identity and empirical heptad scores of the peptide pairs. We reassessed the correlation between the predicted and observed cross-reactivity from the single peptide vaccines based on the revised criteria and observed that the MCC increased significantly from 0.42 to 0.74 (Supplemental Table III).
Applying the new criteria predicting cross-reactive immunogenicity to the design of a next-generation vaccine with high theoretical global coverage
For all combinations of N-terminal M peptide pairs derived from the 117 epidemiologically relevant M types (17), we identified 222 unique pairs of M types that share either ≥45% sequence identity in their 50-mers or ≥40% sequence identity in their overlapping coiled-coil domains. Based on the revised cross-reactivity prediction algorithm, we determined that 69 of the 222 pairs are predicted to demonstrate immunological cross-reactivity. After optimizing to select the minimal number of M types for inclusion in a multivalent vaccine from the 69 cross-reactive pairs that would cover the maximum number of M types, we found that 25 M types are predicted to cross-react with 48 heterologous M types. As a result, with the addition of five highly prevalent type-specific M peptides that are not predicted to cross-react with any other M types (7), a cross-protective multivalent vaccine could theoretically achieve >78% global coverage of Strep A infections, with regional coverage ranging from 63–92%. Future studies will focus on designing and testing these highly complex and potentially broadly protective M protein–based vaccines.
Discussion
The world needs a safe and effective vaccine to reduce the significant global burden of disease caused by Strep A infections and their complications (32). There are two general approaches to the development of Strep A vaccines: those formulated with common protective Ags shared by many or all M types of Strep A and multivalent M protein–based vaccines containing multiple different N-terminal M peptides (33). The N terminus of the M protein contains epitopes that elicit Abs with the greatest bactericidal activity and are least likely to elicit autoimmune Abs (15). Three multivalent M protein–based vaccines containing 6, 24, or 30 N-terminal M peptides have been evaluated in a total of four early-stage clinical studies (10, 12, 24). All three vaccines were well tolerated and immunogenic and elicited functional opsonic Abs in the absence of host tissue cross-reactive Abs.
Among the perceived limitations to the development of multivalent M protein–based vaccines is potential vaccine coverage in low- and middle-income countries and in disadvantaged populations in high-income countries in which the diversity and complexity of M types is high and in which the risk of acute rheumatic fever and rheumatic heart disease is greatest (34–36). The 30-valent vaccine was originally designed based on the prevalence of M types and the epidemiology of infections in North America and Western Europe. Studies in animals showed that the vaccine not only elicited bactericidal Abs against all of the vaccine M types but also cross-opsonic Abs against a significant number of nonvaccine types, suggesting the presence of cross-reactive protective epitopes among heterologous M types of Strep A (15). Subsequent studies revealed that the majority of M proteins could be grouped into sequence-based clusters that were immunologically and functionally related (7). Although there are now >200 different M types of Strep A, which are defined by different N-terminal M protein sequences, there is recent evidence that immunity to Strep A may be a combination of cluster-specific and type-specific M Abs (37, 38). An emerging concept is that the N terminus of the M protein is structurally constrained based on the requirement for binding host proteins that promote virulence. This concept has recently been supported by studies revealing common C4BP motifs contained in seemingly disparate amino acid sequences (19).
Our hypothesis is that the structural constraints placed on the N terminus of M proteins may be exploited to identify cross-reactive and cross-protective epitopes that are hidden within the variable sequences. Most of the M proteins display a coiled-coil structure that extends into the hypervariable N-terminal region. This structural similarity can be used to predict cross-reactivity based on both conserved sequence (39) and structural patterns (16) within M peptides selected as vaccine Ags, potentially resulting in broadly protective immune responses. Although experimental assays can identify immunological cross-reactions among heterologous M peptides, screening every possible cross-reaction among 117 globally prevalent M types would require 6786 pairs. We reasoned that in silico prediction and validation could significantly reduce the experimental burden needed to identify potential vaccine candidates while also defining the structural basis for cross-reactive epitopes. In this study, we have derived a significantly improved algorithm that combines structure- and sequence-based analyses to efficiently identify M peptides that were predicted to cross-react with high specificity and demonstrate its precision in predicting the cross-reactive potential between any M peptide pair.
Using helical wheel alignment, the heptad score was used to determine the degree of structural and chemical similarity at the corresponding polar heptad sites between any M peptide pair. Combining sequence identity with heptad scores in a two-step approach allowed assessment of potentially shared conformational epitopes between any M peptide pair and thus increased the specificity of cross-reactivity prediction compared with sequence identity thresholds alone. A six-valent vaccine comprised of M types identified by the two-tiered approach exhibited complete cross-reactivity and cross-protection against an N-terminal sequence-similar cluster (NTC5) containing 11 M types. All NTC5 M peptides were predicted to have long and well-ordered coiled-coil domains. The two-tiered approach showed excellent specificity for M peptides predicted to have long and ordered coiled-coil domains. However, an analysis of the 117 N-terminal M peptides from epidemiologically significant group A streptococci indicated that 15% of the peptides display short coiled-coil domains and/or low coiled–coil propensity, suggesting that sequence identity and heptad identity scores alone would be insufficient to predict cross-reactivity among the majority of M types. Therefore, we performed additional experiments to test the hypothesis that factoring in coiled-coil propensity of the peptides would improve the positive predictive power of the algorithm. We immunized animals with four different M peptides that were not sequence related. Two of the peptides (M70 and M121) were calculated to have short and low propensity coiled-coil domains, whereas the other two (M67 and M117) were calculated to have long coiled-coil domains. We found that the two peptides with short coiled-coil domains and propensities were immunogenic but elicited Abs that cross-reacted poorly or not at all with many of the M peptides with which they shared sequence identity and heptad scores that were above the threshold. As such, these M peptides would not be considered ideal vaccine candidates. The revised algorithm that specifies different thresholds for M peptide pairs with different levels of average coiled-coil probability and coiled-coil lengths substantially improved the predictive power of the algorithm (MCC of 0.74) and provided a rational explanation for the results in terms of three-dimensional structure.
This rapid and accurate method for predicting cross-reactivity among N-terminal M peptides may facilitate the formulation of vaccines that could potentially induce broadly cross-reactive and cross-protective immunity against the majority of epidemiologically relevant strains of Strep A. When the new algorithm was used to analyze 117 sequence-related globally prevalent M types, we identified 25 M peptides that could potentially cross-react with an additional 48 M types and thereby provide ∼60% potential coverage of Strep A infections globally. The algorithm was designed to have high specificity while sacrificing some sensitivity. Thus, we believe that the actual number of cross-reactive pairs will be much higher. With the addition of five N-terminal peptides from M types that are highly prevalent but are not sequence-related to others, potential global coverage increased to >78%. In summary, our study confirms that the sequence- and structure-based algorithm has the potential to identify vaccine M types that will elicit cross-reactive Abs that promote opsonization of nonvaccine M types. It also answers the question of how many M peptide vaccine targets may be necessary to achieve desirable global coverage of prevalent M types of Strep A. Future studies in animals will be designed to evaluate the breadth of vaccine-specific and cross-reactive immunogenicity of the new vaccine constructs.
Our results may also have broader implications for vaccine design targeting other human pathogens. Immunogenic α-helical protein domains are common among pathogenic bacteria, viruses, and parasites. The search for cross-protective or universal vaccines is an active topic of investigation. Examples include Plasmodium vivax and P. falciparum vaccines (40), S. pneumoniae (PspA) (41), schistosomes (42), the long α-helix of influenza hemagglutinin (43), and protective Ags of Mycobacterium tuberculosis (44). Applying our structure-based approach to this common protein motif may improve the predictions of other investigators searching for broadly protective vaccine Ags.
Acknowledgements
We appreciate the expert technical assistance of Gwenaelle Botquin in performing the whole blood opsonophagocytic killing assays.
Footnotes
This work was supported by National Institutes of Health, National Institute of Allergy and Infectious Diseases Grant R01AI132117.
M.P.A., J.C.S., and J.B.D. conceptualized the project. M.P.A. performed the calculations, collected data, and analyzed the data. T.A.P. and S.S. performed the experiments and collected experimental data. P.S. and A.B. contributed to the whole blood opsonophagocytic killing assays. M.P.A. wrote the manuscript with input from J.B.D. and J.C.S. All authors discussed the data and analysis. J.B.D., J.C.S., and P.S. provided resources and supervised the project.
The online version of this article contains supplemental material.
References
Disclosures
J.B.D. is the inventor of certain technologies related to the development of group A streptococcal vaccines. The technology has been licensed from the University of Tennessee Research Foundation to Vaxent, LLC, of which J.B.D. is a member and the Chief Scientific Officer. The other authors have no financial conflicts of interest.
